

Основные этапы патогенеза атеросклероза
До появления морфологических изменений во внутренней оболочке артерий возникает атерогенная дислипопротеинемия, гиперхолестеринемия с уровнем холестерина свыше 250–300 мг%. Появляются модифицированные липопротеины, происходит их интенсивный захват ЛПОНП-рецепторами и скевенджер-рецепторами эндотелиоцитов.
Начало образования атеросклеротической бляшки и атеросклероза как болезни — повреждение эндотелия, повышение его проницаемости. В результате во внутреннюю оболочку артерий проникает большое количество липопротеинов и других компонентов плазмы крови. Развивается субэндотелиальный мукоидный отёк. После слущивания части повреждённых эндотелиоцитов возможен контакт тромбоцитов с базальной мембраной внутренней оболочки, часть эндотелиальных клеток теряет антикоагулянтные свойства.
Повреждённые эндотелиоциты выделяют адгезивные молекулы (ICAM-1, VCAM-1, LFA-1), что приводит к прилипанию к эндотелию тромбоцитов, моноцитов, лимфоцитов. Тромбоциты выделяют трансформирующий фактор роста (ТФР). Моноциты проникают в субэндотелиальное пространство и превращаются в макрофаги, синтезирующие цитокины (ИЛ-1, ФНО, ТФР, фактор роста фибробластов и др.). Последние вызывают хемотаксис и клеточную пролиферацию. Цитокины лимфоцитов также обеспечивают хемотаксис клеток, участвующих в иммунном воспалении.
Макрофаги, тромбоциты, повреждённый эндотелий выделяют ТФР, стимулирующий гладкомышечные клетки стенки артерий. Гладкомышечные клетки мигрируют во внутреннюю оболочку, где начинают синтезировать протеогликаны, коллаген, эластин, необходимые для построения коллагеновых и эластических волокон. При этом синтезируемые типы коллагена обеспечивают сродство липопротеинов к скоплениям гладкомышечных клеток, это также способствует накоплению липидов.
Во внутренней оболочке артерий происходит пероксидация липопротеинов под влиянием макрофагальных цитокинов, Возникают комплексы липопротеинов с протеогликанами, происходит захват последних макрофагами и гладкомышечными клетками. При этом в миоцитах возможно нерегулируемое поглощение модифицированных ЛПОНП. В макрофагах и гладкомышечных клетках истощается система утилизации, прежде всего, лизосом. Когда цитоплазма загружена липидами, возникают ксантомные клетки. В дальнейшем происходит разрушение макрофагов, гладкомышечных и ксантомных клеток, что способствует накоплению экстрацеллюлярных липидов.
По мере прогрессирования заболевания в атеросклеротической бляшке образуются сосуды, нарастают процессы склероза и гиалиноза, происходит некроз центра бляшки и её обызвествление (рис. 10-1).
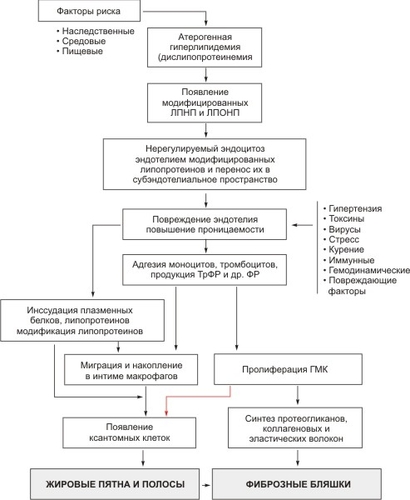
Рис. 10-1. Патогенез атеросклероза. Сокращения: ФР — фактор роста, ТрФР — фактор роста из тромбоцитов, ГМК — гладкомышечная клетка.
В пато- и морфогенезе атеросклероза выделяют несколько стадий: долипидную, стадии липоидоза, липосклероза и осложнённых поражений. Существуют макроскопическая и микроскопическая классификации этих стадий, разница между ними в том, что микроскопическая классификация включает на одну стадию больше. Это так называемая долипидная стадия атеросклероза, когда описанные выше изменения предшествуют накоплению липидов во внутренней оболочке, видному невооружённым глазом. Названия отдельных стадий отражают одни и те же процессы, происходящие в атеросклеротической бляшке.
● Долипидная стадия не имеет клинической симптоматики. Характерны прогрессирующая гиперлипидемия, гиперхолестеринемия, дислипопротеинемия, гипофосфатемия и другие изменения, отражающие нарушения метаболизма при атеросклерозе. Вследствие того, что внутренняя и часть средней оболочки крупных и средних артерий получают питание за счёт диффузии из кровотока, все изменения в крови ведут к морфологическим изменениям внутренней оболочки. В начале происходит повышение проницаемости эндотелия (выраженный пиноцитоз эндотелиоцитов, накопление в них липидных капель, исчезновение гликокаликса, раскрытие межэндотелиальных стыков). Пролиферация гладкомышечных клеток, синтезирующих протеогликаны, способствует подэндотелиальному мукоидному отёку внутренней оболочки, дистрофии и слущиванию эндотелиоцитов. Накопление во внутренней оболочке белков плазмы, гликозаминогликанов создаёт благоприятные условия для фиксации ЛПНП, ЛПОНП, холестерина, апо-β-липопротеинов, фибриногена и др.
◊ Уже на этой стадии во внутренней оболочке происходит трансформация моноцитов в мононуклеарные фагоциты. Они захватывают ЛПНП, транспортируют их во внутреннюю оболочку, секретируют большое количество цитокинов, в том числе, ТФР, способствующий пролиферации гладкомышечных клеток. Макрофаги выделяют также коллагеназу, эластазу, расщепляющие коллагеновые и эластические волокна, липопротеинную липазу, обеспечивающую распад липопротеинов и их фагоцитоз. Кроме того, макрофаги выделяют свободно-радикальный кислород, участвующий в окислении липидов, активной утилизации липопротеинов, образовании иммунных комплексов. Цитокины моноцитов обеспечивают миграцию во внутреннюю оболочку артерий T- и B-лимфоцитов, взаимодействующих с поверхностными антигенами макрофагов, гладкомышечных клеток, эндотелиоцитов.
◊ Таким образом, иммунные реакции предшествуют макроскопическим изменениям во внутренней оболочке. Очевидно, долипидная стадия продолжается до тех пор, пока функционируют системы, обеспечивающие выведение из оболочки артерий липопротеинов и других метаболитов. После истощения этих систем процесс переходит в следующую стадию, когда прогрессирующие морфологические изменения видны невооружённым глазом.
● Стадия липоидоза, или жировых пятен и полосок (рис. 10-2). В эту стадию происходит очаговая инфильтрация внутренней оболочки артерий холестерином, липопротеинами, белками плазмы, моноцитами, макрофагами, гладкомышечными и ксантомными клетками. Выражены набухание и деструкция эластических мембран. Очаги атеросклероза имеют жёлтый или серо-жёлтый цвет, не возвышаются над поверхностью внутренней оболочки, но хорошо видны (рис. 10-3). Они получили название жировых пятен и полосок, возникают в первую очередь на задней стенке аорты, в местах отхождения её ветвей, затем в крупных артериях.
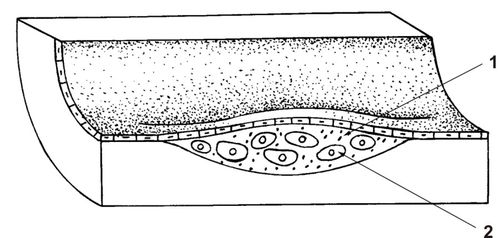
Рис. 10-2. Атеросклеротические изменения в артерии: 1 — межклеточные липиды, 2 — пенистые клетки.
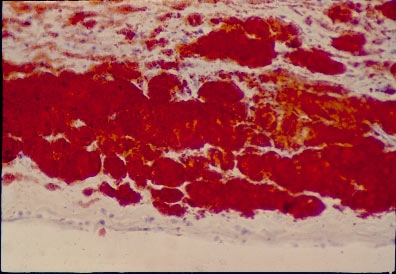
Рис. 10-3. Липоидоз интимы артерии. Окраска суданом III (лупа).
Образования, схожие с жировыми пятнами и полосками, но расположенные в поперечном направлении относительно продольной оси аорты и крупных артерий, получили название поперечных полосок. Они бывают у новорождённых, содержат скопления гладкомышечных клеток, соединительную ткань, но не содержат липиды. В детском и юношеском возрастах поперечные полоски исчезают, однако в последующем часто образование атеросклеротических бляшек на месте поперечных полосок. Поэтому предполагают, что со временем возможна трансформация части поперечных полосок в атеросклеротические бляшки.
● Стадия липосклероза, или фиброзных бляшек начинается с разрастания во внутренней оболочке вокруг очага отложения липидов, липропротеидов, холестерина соединительной ткани. При этом происходит пролиферация гладкомышечных клеток и макрофагов, присутствуют T- и B-лимфоциты, плазматические и ксантомные клетки. Бляшки становятся плотными, белого или бело-жёлтого цвета, овальной или округлой формы, выступают в просвет сосуда (рис. 10-4). В аорте и крупных артериях часто сливание бляшек друг с другом, что придаёт внутренней поверхности сосудов бугристый вид. Верхний фиброзный слой бляшки, обращённый в просвет сосуда и покрытый дистрофически изменённым эндотелием, носит название покрышка бляшки. В краях бляшки происходит новообразование сосудов (vasa plaquorum), через них также поступают липопротеины и плазменные белки, способствуя росту фиброзных бляшек. При увеличении в размерах они сужают просвет артерии.
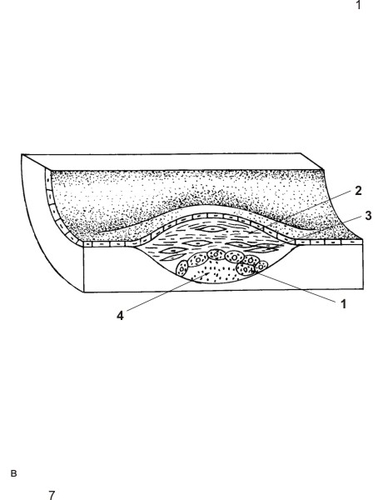
Рис. 10-4. Атеросклеротические изменения в артерии. Фиброзная бляшка: 1 — пенистые клетки, 2 — фиброзная капсула, 3 — гладкомышечные клетки, 4 — липидное ядро.
Фиброзные бляшки — основная форма атеросклеротического поражения сосудов. Их находят в участках артерий, испытывающих повышенное гемодинамическое воздействие (места ветвления и изгибов сосудов, сторона артерий, прилежащая к плотным образованиям, например, задняя стенка аорты, прилежащая к позвоночнику). Бляшки чаще расположены в области дуги и брюшном отделе аорты, артериях сердца, мозга, почек, нижних конечностей, сонных артериях и др.
● Стадия осложнённых поражений включает атероматоз, изъязвление и кальциноз фиброзной бляшки (рис. 10-5).
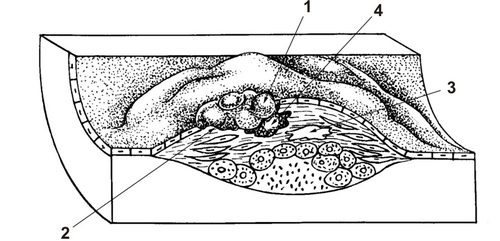
Рис. 10-5. Атеросклеротические изменения в артерии. Стадия осложнённых поражений: 1 — тромб, 2 — изъязвление, 3 — кальцификация, 4 — кровоизлияние.
◊ Атероматоз. Характерны омыление и распад липидов с образованием кристаллов холестерина в центре бляшки, разрушение прилежащих коллагеновых и эластических волокон. В результате образуется атероматозный кашицеобразный детрит. Вокруг него в бляшке расположены ксантомные клетки, активные T-лимфоциты, фрагменты циркулирующих иммунных комплексов, плазматические клетки. Покрышка бляшки нередко гиалинизирована. Средняя оболочка артерий под бляшкой атрофична, что способствует образованию аневризм. В бляшке много vasa plaquorum.
◊ Изъязвление возникает после кровоизлияния в атероматозные массы атеросклеротической бляшки с образованием интрамуральной гематомы. При этом происходит разрыв покрышки бляшки, тромботические и атероматозные массы выпадают в просвет сосуда и могут стать источником тромбоэмболии. Бляшка имеет вид атероматозной язвы с подрытыми, неровными краями и дном, образованным мышечной тканью или наружной оболочкой сосуда. Дефект внутренней оболочки в области атероматозной язвы часто прикрыт тромботическими массами.
◊ Кальциноз завершает морфогенез атеросклеротической бляшки осаждением в ней солей кальция. Происходит обызвествление, или петрификация бляшки. Она приобретает каменистую плотность и состоит из плотных, ломких пластинок.
Нередко сочетание в одном сосуде различных стадий атеросклероза, что указывает на волнообразное прогрессирующее течение заболевания. Все описанные изменения фиброзных бляшек приводят к стенозированию, тромбозу и окклюзии артерий. С этим связаны клинико-анатомические формы атеросклероза. Клинические проявления зависят от локализации и распространённости процесса. При этом медленное стенозирование сосуда атеросклеротической бляшкой вызывает нарастающую ишемию ткани, её атрофию, склероз и хроническую сосудистую недостаточность. Развитие коллатерального кровообращения может привести к перераспределению крови в смежных сосудистых бассейнах и появлению синдромов обкрадывания. При этом компенсация кровоснабжения тканей в районе стенозированной артерии происходит за счёт ухудшения кровоснабжения близлежащих органов. Быстрая окклюзия питающей артерии обычно связана с осложнённым течением атеросклеротической бляшки и приводит к острой недостаточности кровоснабжения, развитию инфаркта или гангрены органа. Кроме того, образовавшаяся атеросклеротическая аневризма может вызвать расслаивание и разрыв стенки артерии с последующим массивным кровотечением.
ОСНОВНЫЕ ФОРМЫ АТЕРОСКЛЕРОЗА
Содержание раздела «Основные формы атеросклероза» смотрите в книге.
ОСЛОЖНЕНИЯ И ИСХОД АТЕРОСКЛЕРОЗА
Исход атеросклероза, как правило, — смерть в результате осложнений заболевания — хронической ишемической болезни сердца, инфаркта миокарда, инсульта головного мозга, гангрены кишечника или нижних конечностей, разрыва аневризмы аорты, кровоизлияний и кровотечений. Исход зависит также от локализации поражения, калибра поражённой артерии и остроты процесса.
АРТЕРИАЛЬНАЯ ГИПЕРТЕНЗИЯ
Гипертензия, или гипертония — симптом повышения артериального давления (АД) при различных заболеваниях, исчезающий после их излечения (симптоматические, или вторичные артериальные гипертензии). Если причина гипертензии неизвестна и повышение АД первичное (идиопатическое), говорят об эссенциальной гипертензии. В России её называют гипертонической болезнью. Нормальным АД, по рекомендации ВОЗ, принято считать АД ниже 140/90 мм рт.ст. АД выше этих цифр расценивают как гипертензию.
ГИПЕРТОНИЧЕСКАЯ БОЛЕЗНЬ
Гипертоническая болезнь (эссенциальная гипертензия, первичная артериальная гипертензия) — хроническое заболевание, его сущность — повышение АД. Длительный и постепенный рост давления крови в сосудах характерен для доброкачественной формы заболевания. Быстрый и высокий подъём АД называют гипертоническим кризом. Если гипертонические кризы повторяются часто, а диастолическое АД выше 120 мм рт.ст., то больной может погибнуть через 2–5 лет заболевания. Такую форму болезни называют злокачественной гипертензией.
Распространённость
Гипертонической болезнью страдает 20–30% взрослого населения мира. Мужчины болеют несколько чаще женщин, сельские жители страдают гипертонической болезнью в 4–5 раз реже жителей крупных городов. Представители негроидной расы заболевают в 2 раза чаще и имеют худший прогноз болезни. Заболевание обычно развивается после 35–45 лет, прогрессирует до возраста 55–58 лет, после чего часто бывает стабилизация на повышенных цифрах АД. Иногда стойкое и быстрое нарастание АД обнаруживают у молодых лиц.
Симптоматические гипертензии составляют 5–10% всех случаев повышения АД, остальные состояния с повышением АД относят к гипертонической болезни.
Гипертоническая болезнь, наряду с ИБС и цереброваскулярными заболеваниями, — основная причина смерти населения развитых стран. В России смертность пациентов с гипертонической болезнью в возрасте 35–65 лет в 2 раза выше, чем в Западной Европе.
Основные факторы, определяющие уровень АД, — сердечный выброс и общее периферическое сосудистое сопротивление. Выделяют факторы риска, влияющие на эти параметры. Большинство из них совпадает с факторами риска развития атеросклероза.
● Генетические факторы (гипертоническая болезнь часто имеет семейный характер и полигенную форму наследования).
● Эмоциональные стрессы, особенно длительные нервно-психические расстройства.
● Избыточное потребление поваренной соли, обычно на фоне генетической предрасположенности к гипертонической болезни. Кроме того, повышенное содержание в пище натрия часто сопровождается недостатком калия, кальция и магния, что играет роль в патогенезе гипертонической болезни.
● Гормональные факторы — усиление прессорных влияний гипоталамо-гипофизарной системы, избыточное выделение катехоламинов, активация рениновой гипертензивной системы.
● Почечный фактор имеет большое значение. При высокой активности ренин-ангиотензин-альдостероновой системы развивается вазоконстрикторная гипертензия, при низкой активности — гиперволемическая гипертензия.
● Расовые факторы (чернокожие мужчины умирают от гипертонической болезни в 6 раз чаще, чем белые).
● Избыточная масса тела.
● Курение и злоупотребление алкоголем.
● Профессиональные факторы: чрезмерное длительное напряжение внимания, сдерживание отрицательных эмоций (например, учителями, преподавателями, врачами), вибрация, электромагнитное поле, постоянный шум и т.п.
● Малоподвижный образ жизни.
Этиология
Гипертоническая болезнь — многофакторное заболевание, вызванное сочетанием генетической предрасположенности и воздействия внешней среды. Основные причины гипертонической болезни:
хроническое психо-эмоциональное перенапряжение — повторные стрессы, конфликтные ситуации, длительное чрезмерное напряжение внимания (теория Г.Ф. Ланга и А.Л. Мясникова);
генетический дефект почечно-объёмного механизма регуляции АД (теория A. Guyton);
генетический дефект ионных насосов клеточных мембран, приводящий к нарушению обмена ионов кальция и натрия (теорияЮ.В. Постнова и С.Н. Орлова).
Патогенез
Условно выделяют нейрогуморальные и гемодинамические механизмы, ведущие к повышению АД.
● Нейрогуморальные факторы повышают тонус периферических артериол и запускают механизмы второй группы.
◊ Симпато-адреналовая система, регулирующая содержание и активность катехоламинов — адреналина, норадреналина, дофамина, эндорфинов и других опиатов.
◊ Ренин-ангиотензин-альдостероновая система.
◊ Гормоны (АКТГ, кортизол, соматотропный гормон, вазопрессин, половые гормоны и др.).
◊ Прессорно-депрессорные системы (простагландиновая и калликреин-кининовая).
◊ Система циклических нуклеотидов.
◊ Изменение транспорта ионов кальция, натрия, калия (наследственный дефект клеточных мембран).
◊ Допаминовая система почек.
◊ Серотониновый механизм.
● Гемодинамические механизмы. Повышение тонуса периферических артериол возникает под влиянием следующих факторов.
◊ Задержка воды и ионов натрия в стенке сосудов.
◊ Прямое вазоконстрикторное действие ангиотензина II, кортизола, простагландина F2α, цГМФ.
◊ Повышение уровня кальция в клетках, особенно в гладкомышечных клетках артериол.
◊ Снижение депрессорных влияний, в результате падает синтез простагландинов Е, А, D, простациклина, брадикинина.
Названные факторы (рис. 10-6) создают гемодинамическую основу гипертонической болезни — неадекватное повышение периферического сопротивления, обусловленное повышением сосудистого давления, увеличением объёма циркулирующей крови и интерстициальной жидкости. У больных гипертонической болезнью повышение сосудистого тонуса всегда не соответствует изменению сердечного индекса.
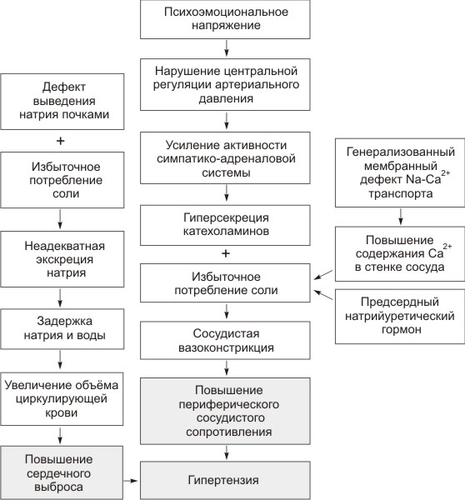
Рис. 10-6. Патогенез артериальной гипертензии.
Для возникновения артериальной гипертензии необходимо повышение сердечного выброса и увеличение общего периферического сопротивления или сочетание этих факторов. Разнообразные механизмы регуляции АД участвуют в патогенезе артериальной гипертензии не одновременно. В начале болезни механизмы повышения АД нейтрализуются механизмами, нормализующими его. Взаимовлияние этих механизмов определяет развитие заболевания (рис. 10-6).
Начало гипертонической болезни — длительное психо-эмоциональное перенапряжение, приводящее к снижению тормозного влияния коры головного мозга на подкорковые центры и появлению застойного очага возбуждения в гипоталамо-гипофизарной системе (Н.Ф. Ланг, А.Л. Мясников, П.К. Анохин). Это нарушает регуляцию подкорковых вегетативных центров и жиро-белкового обмена. Возникает стойкое перевозбуждение прессорных центров и, как следствие, — спазм артериол и повышение АД. Это стимулирует барорецепторы каротидного синуса и дуги аорты, что на начальных этапах заболевания нормализует АД за счёт стимуляции вазомоторного центра продолговатого мозга. При сохранении психо-эмоционального перенапряжения нарастает нагрузка на барорецепторы синокаротидной зоны. К нарушению центральной регуляции вазомоторных влияний присоединяется активация симпато-адреналовой системы с увеличением в крови уровня катехоламинов, также способствующих подъёму АД. Спазм артериол почек в сочетании с симпатическими влияниями включает почечный фактор (прежде всего, почечно-объёмный механизм регуляции АД, а через юкстагломерулярный аппарат — включение ренин-ангиотензин-альдостероновой системы, усиливающей спазм артериол).
Участие в патогенезе болезни почечного фактора связано с избыточным потреблением соли. Длительному спазму артериол способствует генетический дефект клеточных мембран, в том числе, мембран миоцитов, приводящий к накоплению в клетках ионов кальция и натрия. Это делает их особо чувствительными к прессорным влияниям нейромедиаторов и других гуморальных факторов.
Реакция почечно-объёмного механизма на повышение АД — снижение экскреции ионов натрия, что ведёт к задержке в организме воды и натрия, в том числе, в гладкомышечных клетках стенок артериол, увеличению объёма плазмы крови и межтканевой жидкости. Возникает и прогрессирует гиперволемия, возрастают венозный возврат и сердечный выброс. Задержка натрия и воды повышает тонус сосудов, их чувствительность к прессорным факторам, что также способствует повышению АД. Увеличение объёма плазмы крови, в свою очередь, усиливает секрецию натрийуретического гормона, ингибирующего Na+,K+-зависимой АТФазы не только в почках, но и во всём организме. Это увеличивает экскрецию натрия и воды почками, не снижая гиперволемии. Кроме того, натрийуретический гормон задерживает натрий и воду в эритроцитах и стенках артериол, что повышает сосудистое сопротивление.
Увеличение АД до определённого уровня устанавливает новое равновесие между поступающей и экскретируемой солью, прекращая задержку воды. Также происходит «перенастройка» (по П.К. Анохину) барорецепторов на новый, повышенный уровень АД. Возникает замкнутый круг, в результате АД возрастает в течение длительного времени.
Нарушение регуляции жиро-белкового обмена проявляется накоплением в крови ЛПНП и ЛПОНП, снижением содержания ЛПВП и ЛПОВП. Это влияет на механизмы регуляции АД, а постоянное повышенное АД усиливает прогрессирование атеросклеротического процесса. Изменение липидного состава плазмы крови может влиять на липопротеинный комплекс мембран клеток. Доказана зависимость между артериальной гипертензией и патологией липидов клеточных мембран, что нарушает функцию катионных помп, транспорт ионов кальция, натрия и других катионов. В ответ на длительную нагрузку повышенным давлением в стенках аорты, крупных и средних артерий возникает гиперэластоз, позже эластофиброз и повреждение эндотелия. Таким образом, развивается характерный для гипертонической болезни артериосклероз. Постоянное действие повреждающих факторов ведёт к большей выраженности гипертонической макроангиопатии, чем при атеросклерозе. Атеросклероз артерий, вызывающий ригидность стенок артерий, атеросклеротическое разрушение барорецепторов, также участвует в патогенезе гипертонической болезни.
Очевидно, возникновение и патогенез гипертонической болезни зависят от сочетания основных факторов: хронического психо-эмоционального перенапряжения, генетического дефекта клеточных мембран, ведущего к нарушению обмена ионов кальция и натрия, и генетических нарушений почечно-объёмного механизма.
Существуют доброкачественная и злокачественная формы гипертонической болезни.