

2.2. Квантовомеханический случай
Совсем по-иному подходит квантовая механика для нахождения энергии жесткого ротатора. Общий квантовомеханический подход для нахождения энергии заключается в решении уравнения Шредингера для жесткого ротатора
![]() ,
(2.7)
,
(2.7)
где
![]() –
оператор Гамильтона для жесткого
ротатора, ψвр
–
вращательная волновая функция, Евр
– собственное
значение энергии оператора.
–
оператор Гамильтона для жесткого
ротатора, ψвр
–
вращательная волновая функция, Евр
– собственное
значение энергии оператора.
Учитывая,
что для жесткого ротатора можно считать
его потенциальную энергию V
= 0,
оператор Гамильтона будет равен только
оператору кинетической энергии
![]() такой системы. Уравнение Шрёдингера
для стационарных состояний жесткого
ротатора формально будет совпадать с
уравнением движения свободной частицы
с кинетической энергией Т
и массой m:
такой системы. Уравнение Шрёдингера
для стационарных состояний жесткого
ротатора формально будет совпадать с
уравнением движения свободной частицы
с кинетической энергией Т
и массой m:
![]() ,
(2.8)
,
(2.8)
где
![]() –
оператор кинетической энергии частицы,
–
оператор кинетической энергии частицы,
![]() –
оператор Лапласа. символ
(набла
в квадрате) означает оператор, определяемый
соотношением
–
оператор Лапласа. символ
(набла
в квадрате) означает оператор, определяемый
соотношением
![]() .
Для вращающегося
ротатора это уравнение можно переписать
следующим образом:
.
Для вращающегося
ротатора это уравнение можно переписать
следующим образом:
![]() ,
(2.9)
,
(2.9)
где приведенная масса вращающейся молекулы. Если это уравнение переписать в сферических координатах r, , связанных с декартовыми координатами x, y, z соотношениями x = rsin sin, y = rsincos, z = rcos, то оно примет вид:
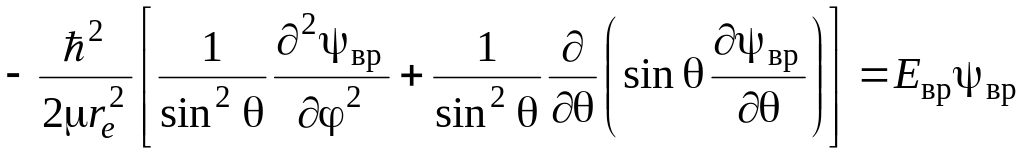 .
(2.10)
.
(2.10)
Здесь учтено то обстоятельство, что из общего выражения Лапласиана
![]() ,
,
первый
член равен нулю, так как радиальная
координата не изменяется в процессе
вращения (жесткий ротатор).
поэтому
вращательная волновая функция
![]() зависит
только от угловых координат
и ,
а значение
r
равно
.
Таким образом,
вращение двух масс m1
и m2
вокруг одной из осей, проходящих через
центр тяжести системы, мы заменяем
вращением массы .
(приведенная масса молекулы), находящейся
на расстоянии
от центра сферической системы координат
(рис. 2.3).
зависит
только от угловых координат
и ,
а значение
r
равно
.
Таким образом,
вращение двух масс m1
и m2
вокруг одной из осей, проходящих через
центр тяжести системы, мы заменяем
вращением массы .
(приведенная масса молекулы), находящейся
на расстоянии
от центра сферической системы координат
(рис. 2.3).
Решение уравнения (2.9) можно представить в виде произведения двух функций ( = f()j(), где функции f и j зависят только от одной переменной. Учет граничных условий приводит к тому, что значение энергии Eвр принимает только дискретные собственные значения:
![]() , (2.11)
, (2.11)
где J – вращательное квантовое число, принимающее значение J = 0, 1, 2, 3, …
Выражение
для энергии вращательных состояний
молекулы можно получить гораздо проще,
не решая уравнения Шрёдингера, а приняв
во внимание только квантование момента
вращения
![]() .
.
Согласно законам квантовой механики для свободной системы (в нашем случае двухатомной молекулы) квантуется квадрат момента количества движения. Он принимает ряд дискретных значений, определяемых формулой
![]() , (2.12)
, (2.12)
где J – вращательное квантовое число, принимающее целые значения (J = 0,1, 2, 3, …).
Одновременно квантуется и проекция вращательного момента количества движения на выделенное направление относительно неподвижной (лабораторной) системы координат. Эту систему координат будем обозначать X, Y, Z в отличие от системы координат X, Y, Z, cвязанной с молекулой. Проекция момента вращения относительно неподвижной оси (скажем, оси Z) равна:
![]() , (2.13)
, (2.13)
где mJ = J, J – 1, …, 0,…, –J принимает 2J + 1 значений. Энергия вращающейся молекулы не зависит от проекции момента количества движения, а только от величины квадрата момента, т. е.
![]() (2.14)
(2.14)
Здесь учтено, что P = I.
Так как энергия молекулы не зависит от проекции момента количества движения, а только от квадрата момента, то говорят, что вращательные уровни анергии свободной молекулы всегда вырождены со степенью вырождения g = 2J + 1 (кроме уровня J = 0, для которого g = 1). Это связано с произвольностью ориентации момента количества движения относительно неподвижной системы координат.
Принимая во внимание выражение (2.12) для квадрата момента количества движения, равенство (2.14) можно переписать следующим образом:
![]() ,
(2.15)
,
(2.15)
где
значение момента инерции
![]() относится
к равновесному расстоянию между атомами
(от латинского слова equilibrium
–
равновесие). Как видим, с учетом квантования
квадрата момента количества движения,
мы получили значение вращательной
энергии, как и в (2.11). Согласно выражению
(2.15) коэффициент при вращательном
квантовом числе есть величина постоянная
для данной молекулы. Обозначим этот
коэффициент через
относится
к равновесному расстоянию между атомами
(от латинского слова equilibrium
–
равновесие). Как видим, с учетом квантования
квадрата момента количества движения,
мы получили значение вращательной
энергии, как и в (2.11). Согласно выражению
(2.15) коэффициент при вращательном
квантовом числе есть величина постоянная
для данной молекулы. Обозначим этот
коэффициент через
![]() ,
тогда формулу (2.15) можно переписать
более просто:
,
тогда формулу (2.15) можно переписать
более просто:
![]() ,
(2.16)
,
(2.16)
где
![]() –
вращательная постоянная, относящаяся
к минимуму кривой потенциальной энергии
и соответствующая равновесному расстоянию
между атомами. Эта постоянная полностью
определяет значение вращательной
энергии двухатомной молекулы. При
изучении оптических спектров вращательную
постоянную
обычно выражают в см–1
(в системе СИ она, как и энергия, выражается
в Дж). Оценим для молекул H2
и HCl
величину
в Дж и в см–1.
для
чего будем использовать ряд фундаментальных
постоянных h
= 6,626·10–34Дж·с;
с= 2,998·108
м/с;
I а.е.м. = 1,66·10–27
кг; Н=1,67310–27
кг; Сl35=58,110–27
кг;
reH2 = 0,074 нм;
reHCl =
0,1275
нм.
–
вращательная постоянная, относящаяся
к минимуму кривой потенциальной энергии
и соответствующая равновесному расстоянию
между атомами. Эта постоянная полностью
определяет значение вращательной
энергии двухатомной молекулы. При
изучении оптических спектров вращательную
постоянную
обычно выражают в см–1
(в системе СИ она, как и энергия, выражается
в Дж). Оценим для молекул H2
и HCl
величину
в Дж и в см–1.
для
чего будем использовать ряд фундаментальных
постоянных h
= 6,626·10–34Дж·с;
с= 2,998·108
м/с;
I а.е.м. = 1,66·10–27
кг; Н=1,67310–27
кг; Сl35=58,110–27
кг;
reH2 = 0,074 нм;
reHCl =
0,1275
нм.
Вычислим моменты инерции Ie:
![]()
 Вычислим
в
Дж:
Вычислим
в
Дж:
 .
.
Учитывая,
что величина
![]() постоянна и равна в системе СИ
5,5610–69(Дж)2с2,
то выражение для
постоянна и равна в системе СИ
5,5610–69(Дж)2с2,
то выражение для
![]() будет иметь более простой вид:
будет иметь более простой вид:


Вычислим
в см–1
(так как
![]() ,
то для выражения
в см–1
необходимо полученные величины выражать
в системе СИ за исключением постоянной
с, которую следует выражать в смс–1).
Итак, получим:
,
то для выражения
в см–1
необходимо полученные величины выражать
в системе СИ за исключением постоянной
с, которую следует выражать в смс–1).
Итак, получим:
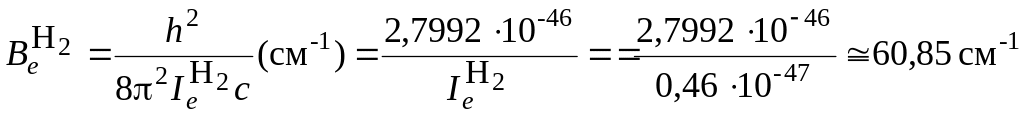
![]() .
.
Из оценки вращательной постоянной видно, что чисто вращательные спектры располагаются в далекой ИК-области, соответствующей сантиметровому и миллиметровому диапазонам.
Так как значения вращательной энергии квадратично зависят от вращательного квантового числа J, то уровни вращательной энергии представляют собой расходящуюся последовательность.
В табл. 2.1 приведены значения Евр в зависимости от вращательного квантового числа J, а в табл. 2.2 межъядерные расстояния двухатомных молекул. Чтобы описать спектр поглощения или испускания, необходимо знать вероятности переходов между уровнями энергии и правила отбора.
Таблица 2.1