

4.4. Малые колебания в квантовой механике
Для определения уровней энергии и волновых функций при колебаниях молекулы необходимо решить уравнение Шредингера вида
![]() .
(4.21)
.
(4.21)
Запись указанного уравнения для i-го нормального колебания будет иметь следующий вид:
 (4.22)
(4.22)
причем
![]() .
(4.23)
.
(4.23)
Формула (4.22) представляет собой известное волновое уравнение для линейного гармонического осциллятора (здесь имеется в виду, что любое i-е нормальное колебание совершается по гармоническому закону). Значения энергии гармонического осциллятора определяются выражением
![]() (4.24)
(4.24)
i = 0, 1, 2, 3, ..., n; i – колебательное квантовое число;i – классическая частота (в с–1) i-го нормального колебания.
Частота света, поглощаемого или излучаемого молекулой, равна
![]() .
(4.25)
.
(4.25)
Переход из основного состояния (все = 0) в состояние, в котором i = 1, a все остальные i(i k) равны нулю, соответствует поглощению частоты k. , являющейся частотой k-го нормального колебания. Согласно правилам отбора для гармонического осциллятора в испускании и поглощении возможны лишь переходы с изменением квантового числа k на ±1.
Таким образом, имеется важное соответствие между результатами квантовой и классической теорий. Для гармонического осциллятора частота квантового перехода между соседними колебательными уровнями в соответствии с правилами отбора совпадает с классической частотой колебаний. Поэтому в гармоническом приближении задача о нормальных колебаниях молекулы может решаться методами классической механики. На рис.4.1 приведены уровни энергии трехатомной молекулы воды.
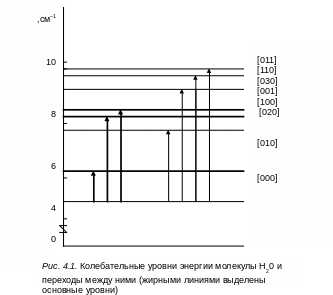
Она
имеет три колебательные степени свободы
и характеризуется тремя нормальными
колебаниями
1=
3657 см–1,
2.=
1595см–1и
3
=
3756 см–1.
Если все i
= 0, то мы,
согласно (4.24), получаем нулевую энергию
колебаний
![]() (где n
= 1, 2, 3),
равную
4504 см
–1.
Основные уровни энергии проведены более
жирными линиями, а обертонные и составные
уровни
– более
тонкими линиями. Переходы на составные
и обертонные уровни возможны при учете
ангармонизма колебаний. Для колебаний
многоатомной молекулы, так же как и для
двухатомной, нужно учитывать
ангармоничность, проявляющуюся тем
сильнее, чем больше колебательная
энергия, приходящаяся на отдельные
нормальные колебания. Ангармоничность
обусловлена отличием реальных сил в
молекуле от квазиупругих. В связи с этим
и потенциальная энергия молекулы не
является чисто квадратичной функцией.
С учетом ангармоничности колебаний
энергия молекулы будет определяться
формулой:
(где n
= 1, 2, 3),
равную
4504 см
–1.
Основные уровни энергии проведены более
жирными линиями, а обертонные и составные
уровни
– более
тонкими линиями. Переходы на составные
и обертонные уровни возможны при учете
ангармонизма колебаний. Для колебаний
многоатомной молекулы, так же как и для
двухатомной, нужно учитывать
ангармоничность, проявляющуюся тем
сильнее, чем больше колебательная
энергия, приходящаяся на отдельные
нормальные колебания. Ангармоничность
обусловлена отличием реальных сил в
молекуле от квазиупругих. В связи с этим
и потенциальная энергия молекулы не
является чисто квадратичной функцией.
С учетом ангармоничности колебаний
энергия молекулы будет определяться
формулой:
![]()
содержащей наряду с линейными членами и квадратичные. величина ангармоничности dik выражается в см–1. Формула (4.26) отличается от аналогичной формулы для двухатомной молекулы тем, что она содержит не только диагональные члены типа dii(i + 1/2)2, но и недиагональные – типа dik(i + 1/2)(k + 1/2), характеризующие взаимное влияние нормальных колебании. При наличии ангармоничности каждое нормальное колебание не только изменяется, но и перестает быть независимыми друг от друга.
коэффициенты
dik
малы по
сравнению с частотами
![]() ,отношение
,отношение
![]() составляет несколько сотых, как и для
двухатомных молекул.
составляет несколько сотых, как и для
двухатомных молекул.
Учет ангармоничности для многоатомных молекул представляет довольно трудную задачу, которая может быть решена для простейших многоатомных молекул. Он наиболее существен для молекул, содержащих атомы водорода (углеводороды, вода и др.).
Вследствие ангармоничности частоты переходов i = 0 i = 1, соответствующих отдельным нормальным колебаниям, не совпадают с нулевыми частотами 0k гармонических колебаний. Нулевая энергия колебаний равна
![]() (4.27)
(4.27)
и частота перехода i = 0i = 1 составляет
![]() (4.28)
(4.28)
В последней формуле (4.28) коэффициент dii характеризует ангармоничность данного колебания, а коэффициенты dij (i j) – связи различных нормальных колебаний, обусловленные ангармоничностью.
Для случая нелинейной трехатомной молекулы частоты переходов будут выражаться следующим образом:
![]() (4.29)
(4.29)
Для молекулы воды разница наблюдаемых i и нулевых частот 0i, оказывается весьма заметной
1 = 3657 см–1, 2 = 1595 см–1, 3 = 3756 см–1,
01 = 3825,3 см–1; 02 = 1653,9 см–1; 03 = 3965,6 см–1.
С учетом ангармоничности правила отбора по колебательному квантовому числу i = ±1 для гармонического осциллятора нарушаются. Оказываются возможными переходы с i 1, которым соответствуют обертоны и составные частоты, получающиеся при одновременном изменении квантовых чисел двух или нескольких колебаний. Поэтому, как видно на рис. 4.1, в молекуле воды наряду с основными переходами (переходы с нулевого колебательного уровня)
[0 0 0] [1 0 0]; [0 0 0] [0 1 0]; [0 0 0] [0 0 1] – 1, 2, 3 основные частоты;
можно наблюдать обертоны:
[2 0 0]; [0 2 0]; [0 0 2]; – первый обертон – 2 1, 2 2, 2 3,
[3 0 0]; [0 3 0]; [0 0 3] – второй обертон –3 1, 3 2, 3 3,
составные частоты:
[1 1 0]; [1 0 1]; [0 1 1] – 1+ 2, 1+ 3, 2+ 3,
[2 1 0]; [2 0 1]; [1 2 0] и др.
Таким образом, получается большое число возможных переходов. На опыте классифицировать основные колебания и их обертоны или составные частоты сравнительно легко, так как интенсивность их значительно меньше основных колебаний.
В спектрах поглощения существенное значение имеют переходы, связанные с основным состоянием, так как при нормальных условиях большинство молекул находится в основном состоянии. Однако могут встречаться и переходы из возбужденных состояний в состояния с большей энергией. соответствующие частоты будут называться разностными частотами.