

4. 5. Классификация нормальных колебаний по форме
Форма
нормального колебания определяется
соотношением амплитуд q1k0,
q2k0,
q3k0,
.... с которыми изменяются колебательные
координаты при k-м
нормальном колебании. Например, для
колебаний молекулы воды форма колебания
будет задаваться соотношением между
величинами амплитуд
![]() ,
,
![]() ,
с которыми изменяются первая и вторая
связь, и угла 0
между ними. На рис.4.2
показано одно из колебаний молекулы
воды. Оно состоит в одновременном
изменении длин обоих связей O–Н
и угла Н–О–Н и характеризуется
амплитудами q10,
q20(q10
= q20)
и 0,
с которыми изменяются координаты
q1,
q2
и .
,
с которыми изменяются первая и вторая
связь, и угла 0
между ними. На рис.4.2
показано одно из колебаний молекулы
воды. Оно состоит в одновременном
изменении длин обоих связей O–Н
и угла Н–О–Н и характеризуется
амплитудами q10,
q20(q10
= q20)
и 0,
с которыми изменяются координаты
q1,
q2
и .
Аналогичным образом оценивают формы колебания в молекулах, содержащих четыре и более атомов. Когда молекула совершает колебание с определенной частотой, то в принципе, в этом колебании участвуют все атомы, т. е. все связи и углы изменяются одновременно. Однако это не значит, что доля участия каждого атома в данном колебательном движении одинакова. Одни связи и углы вовлекаются в это движение более существенно, а другие принимают малое участие. Большей частью встречаются такие случаи, при которых колебательное движение сосредоточено преимущественно в определенной части молекулы. В некоторых предельных случаях можно говорить только об изменении одной связи или величины лишь одного угла. Могут существовать колебания, в которых участвуют все связи при сравнительно неизменных углах между ними, и наоборот, могут изменяться величины углов между связями при неизменном значении длин связей.

Все сказанное позволяет произвести разделение нормальных колебаний (по их форме) на валентные и деформационные.
Валентными будем называть такие колебания, при которых изменяются преимущественно длины связей, а углы между ними остаются постоянными, т. е. qi ≠ 0,
В предельном случае чисто валентных колебаний амплитуда деформационных колебаний точно равна нулю. На рис. 4.3 для молекулы воды показаны чисто валентные колебания, происходящие с частотами 1и 3 и чисто деформационное колебание частоты 2. Это так называемый предельный случай чисто валентных колебаний и чисто деформационных колебаний. При изменении длин связей изменение угла между связями равно нулю и наоборот.
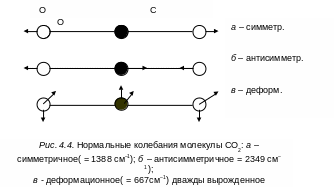
Примером чисто валентных колебаний являются нормальные колебания линейных молекул, при которых атомы движутся вдоль химических связей (колебания молекулы СО2 на рис. 4.4).
Другим примером могут являться чисто валентные колебания (полносимметричного типа) молекулы метана СН4, при которых все атомы водорода колеблются относительно атома углерода вдоль тетраэдрических осей третьего порядка. При этом все четыре связи изменяются одинаковым образом, а углы между связями остаются постоянными (109°28).
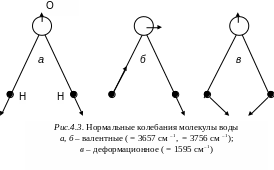
Деформационными будем называть такие колебания, при которых изменяются преимущественно углы между связями, а длины связей остаются приблизительно постоянными, т. е. qi = 0, ≠
Для
молекулы воды чисто деформационным
является колебание
![]() ,
для которого q10
q20
= 0, a
0
.
Указанный предельный случай чисто
деформационных колебаний может
осуществляться для молекул, обладающих
определенной симметрией. Другим примером
деформационного колебания является
случай в) для СО2
на рис.4.4,
при котором атом углерода и атомы
кислорода колеблются с противоположными
фазами перпендикулярно направлению
химических связей в молекуле. Длины
связей при этом остаются почти неизменными.
,
для которого q10
q20
= 0, a
0
.
Указанный предельный случай чисто
деформационных колебаний может
осуществляться для молекул, обладающих
определенной симметрией. Другим примером
деформационного колебания является
случай в) для СО2
на рис.4.4,
при котором атом углерода и атомы
кислорода колеблются с противоположными
фазами перпендикулярно направлению
химических связей в молекуле. Длины
связей при этом остаются почти неизменными.
В ряде молекул может наблюдаться такой случай, при котором заметно изменяются как длины связей, так и углы между ними. Но все же приближенное разделение колебаний на валентные и деформационные сохраняется. Основой такого разделения служит значительное различие силовых постоянных деформационных колебаний по сравнению с силовыми постоянными валентных колебаний. Для многоатомных молекул наряду с классификацией колебаний валентного и деформационного типа важное значение имеет разделение колебаний по степени их локализации в той или иной части молекулы. Встречаются колебания, в которых преимущественно участвуют почти все связи и углы в молекуле (например, скелетные колебания молекулы бензола). Это так называемые делокализованные колебания.
Примером локализованных колебаний могут служить колебания атомов водорода в группах СН2 и СН3, или колебания двойной С=С или тройной связей СС в углеводородах. Локализация колебаний в определенной части молекулы или определенной связи существенно зависит от соотношения между массами колеблющихся атомов. Если между собой связаны два атома различной массы, то в колебаниях относительно общего центра тяжести участвует преимущественно легкий атом. Например, в группах СН, СН2, CH3 валентные колебания С–Н слабо зависят от связи атомов углерода с другими, более тяжелыми атомами. Этот факт является очень важным при определении характеристичности колебаний.
Для грубой оценки частот колебаний многоатомной молекулы можно пользоваться формулой определения частоты гармонических колебаний двухатомной молекулы:
![]() (4.30)
(4.30)
где Кi,.– квазиупругая постоянная, a Mi, – приведенная масса атомов, образующих данную связь (или аналогичная величина для атомов, образующих данный угол).
Пользуясь формулой 4.30, можно найти соотношение между частотами i-го и j-го колебаний:
![]() ,
(4.31)
,
(4.31)
т. е. отношение между частотами колебаний определяется отношением силовых постоянных и приведенных масс. Обычно силовая постоянная К деформационных колебаний много меньше силовой постоянной валентных колебаний. Принимается, что Kдеф примерно в десять раз меньше силовой постоянной валентных колебаний. Это приведет к тому, что при прочих равных условиях согласно (4.31) деф/ вал 1/3. Действительно, частоты деформационных колебаний в два – три раза меньше частот соответствующих валентных колебаний. При сравнении частот различных валентных колебаний одной природы атомов или различных деформационных колебаний главную роль играет различие масс атомов и совсем незначительную – различие силовых постоянных.
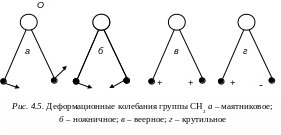
Деформационные колебания характеризуются в свою очередь разнообразием форм движения атомов относительно направления химических связей в молекуле. Различают внутренние и внешние деформационные колебания. Кроме того, различают деформационные колебания отдельных групп атомов относительно других групп или частей молекулы (крутильные, веерные, маятниковые, ножничные и др.).
Например, при внутренних деформационных колебаниях (в группах СН3 или СН2) изменяются углы внутри группы СН3 или СН2. Сам углерод группы CH3 или СН2 может быть соединен одинарной или двойной связью с другим атомом углерода. При внешних деформационных колебаниях С–Н изменяются углы С–С–Н или С=С–Н, определяющие ориентацию всей группы СН3, или СН2 в целом относительно связей С–С или С=С. Другим примером более сложных деформационных колебаний группы СН2 как целого могут быть колебания, приведенные на рис. 4.5.
Малая масса атома водорода по сравнению с массой атомов С, О, N и других приводит к тому, что частоты валентных колебаний С–Н, O–Н, N–H располагаются в области 3000–3500 см–1 (см. характеристические частоты колебаний). Деформационные колебания углов Н–С–Н находятся в области ~ 1400 см –1, а частоты изменения углов С–С–С – 300 – – 400 см –1, т. е. примерно в три раза меньше. Это находится в хорошем соотношении между массами атомов.
Следует отметить, что приведенная классификация колебаний по их форме является приближенной. В реальных ситуациях встречаются промежуточные (между валентными и деформационными) колебания.
Отнесение таких колебаний к определенному типу теряет физический смысл. Однако даже приближенная классификация колебаний, основанная на рассмотрении предельных случаев, имеет большое значение при изучении колебательных спектров, особенно при установлении характеристических частот. Указанные частоты соответствуют колебаниям определенных связей или определенных групп связей в молекуле, которая в равновесном состоянии характеризуется определенным пространственным расположением атомов и химических связей и обладает определенной симметрией.