

Реактивное поле, рассчитанное в модели Онзагера, равно
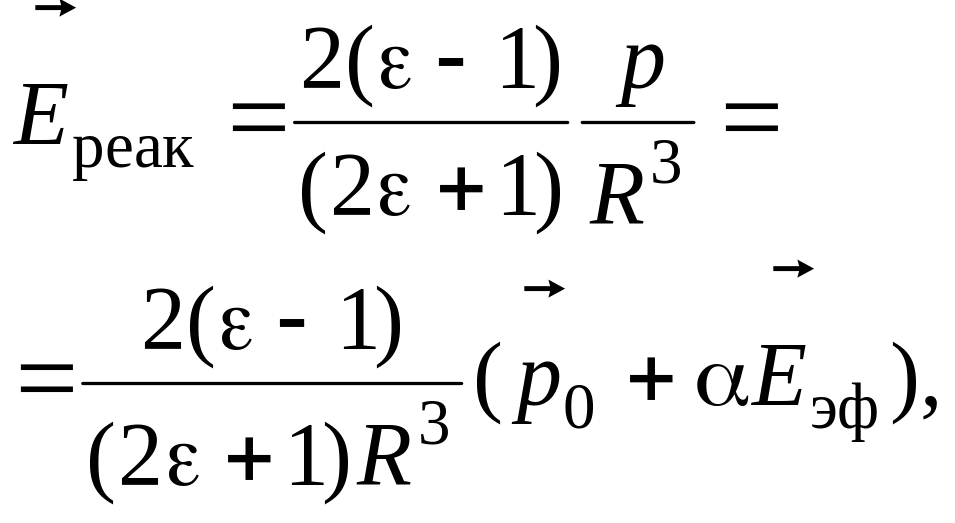 (6.56)
(6.56)
где R – радиус полости, – результирующий дипольный момент молекулы. Реактивное поле, так как и поле внутри полости, можно выразить через показатель преломления среды:
![]() .
(6.57)
.
(6.57)
Таким образом, эффективное поле, действующее на молекулу в озагеровской полости равно
![]()
или
![]() (6.58)
(6.58)
где
![]() и
и
![]() – функции от показателя преломления
среды n
(в общем случае n
может быть комплексным), R
– параметр теории, характеризующий
объем полости.
– функции от показателя преломления
среды n
(в общем случае n
может быть комплексным), R
– параметр теории, характеризующий
объем полости.
Из
формулы (6.58) видно, что свойства
эффективного поля в области примесной
молекулы существенно зависят от ее
поляризуемости и параметра теории R.
Если в единице объема находится N
молекул, которые занимают объем
![]() ,
то R3
определяет эффективный объем, приходящийся
на одну молекулу. Этот объем может быть
рассчитан по известным значениям
плотности и молекулярного веса для
однокомпонентной среды.
,
то R3
определяет эффективный объем, приходящийся
на одну молекулу. Этот объем может быть
рассчитан по известным значениям
плотности и молекулярного веса для
однокомпонентной среды.
Известно,
что объем молекул очень тесно связан с
ее поляризуемостью
,
входящей в формулу Лоренц – Лоренца.
Анализ этой зависимости приводит к
простому выражению
![]() .
.
Следовательно, онзагеровский радиус может быть оценен независимыми способами на основании различных характеристик вещества: показателей преломления или соответствующей величины рефракции, значения которых приведены в справочной литературе. Указанные способы применяются и к двухкомпонентным растворам, если радиусы молекулы и растворителя близки. Если радиус примесной молекулы больше, чем радиус молекулы растворителя, то онзагеровкий радиус для такой системы может быть выбран исходя из ван–дер–ваальсова радиуса исследуемой молекулы.
Модель Онзагера не лишена и некоторых недостатков:
модель шара с точечным диполем в центре не отражает реальных свойств молекулы, обладающей сложным расположением зарядов
нельзя рассматривать ближайшее окружение молекулы как сплошную среду с диэлектрической проницаемостью .
Недостаточно считать, что и связь данной молекулы с ее окружением выражается реактивным полем, имеющим то же направление, что и дипольный момент рассматриваемой молекулы. Логично предположить, что ориентация данной молекулы связана с ориентацией соседних молекул и, следовательно, зависит от их расположения.
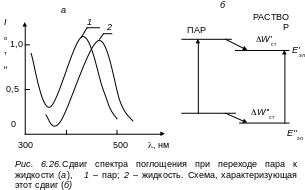
Несмотря на отмеченные недостатки с помощью этой модели можно рассчитывать взаимодействие растворенной молекулы с окружающей средой и определять положение ее электронно-колебательных уровней. Уровень энергии при переходе от газовой фазы к раствору смещается на величину так называемой энергии стабилизации Wст, под которой будем понимать энергию уровня, которая установится после осуществления всех релаксационных процессов в молекуле после акта поглощения или испускания. Разность энергий стабилизации ΔWст = W'ст + W''ст основного и возбужденного уровней молекулы в растворе характеризует смещения спектра поглощения или испускания (см. рис. 6.26)
 Величина
сдвига спектра может быть записана в
следующем виде:
Величина
сдвига спектра может быть записана в
следующем виде:
![]() ,
,
где
![]() ,
,
![]() и
и
![]() – изменения энергии молекулы в растворе,
обусловленные взаимодействиями
ориентационного, индукционного и
дисперсионного характера. Энергия
взаимодействия растворенной молекулы
со средой определяется значениями ее
дипольного момента
,
поляризуемости
,
а также величиной эффективного
электрического поля
,
действующего на молекулу в растворе.
Однако преимущества метода локального
поля проявились еще и в том, что на его
основе были рассчитаны динамические
сдвиги полос в электронных спектрах
поглощения. Влияние динамических
индуктивно-резонансных взаимодействий
на сдвиг частот максимумов полос спектров
растворов был рассмотрен Бахшиевым в
рамках описания общих закономерностей
влияния растворителя на смещение
электронных спектров при переходах от
газовой фазы к раствору. Величина сдвига
оценивается по формуле
– изменения энергии молекулы в растворе,
обусловленные взаимодействиями
ориентационного, индукционного и
дисперсионного характера. Энергия
взаимодействия растворенной молекулы
со средой определяется значениями ее
дипольного момента
,
поляризуемости
,
а также величиной эффективного
электрического поля
,
действующего на молекулу в растворе.
Однако преимущества метода локального
поля проявились еще и в том, что на его
основе были рассчитаны динамические
сдвиги полос в электронных спектрах
поглощения. Влияние динамических
индуктивно-резонансных взаимодействий
на сдвиг частот максимумов полос спектров
растворов был рассмотрен Бахшиевым в
рамках описания общих закономерностей
влияния растворителя на смещение
электронных спектров при переходах от
газовой фазы к раствору. Величина сдвига
оценивается по формуле
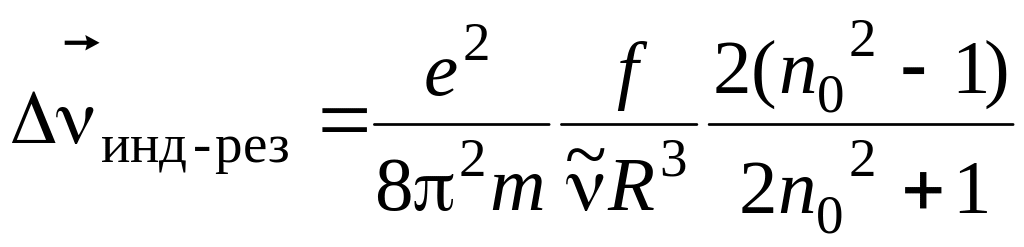 ,
(6.59)
,
(6.59)
где e и m – заряд и масса электрона; f – сила осциллятора полосы; n0 –показатель преломления растворителя; R – онзагеровский радиус молекулы; – частота максимума полосы
Принципиальное отличие динамических сдвигов от статических, рассматриваемых в теории Дебая, состоит в том, что динамические взаимодействия в конечном итоге определяются дипольным моментом перехода между квантовыми состояниями растворенной молекулы.
Итак, полный сдвиг частоты чисто электронного перехода для поглощения выразится в виде суммы
![]() (6.60)
(6.60)
где
![]() –
динамический сдвиг полосы, обусловленной
возникновением у молекулы наведенного
дипольного момента под действием
электрического вектора световой волны.
–
динамический сдвиг полосы, обусловленной
возникновением у молекулы наведенного
дипольного момента под действием
электрического вектора световой волны.
В электронных спектрах поглощения смещение полос может достигать весьма значительной величины, равной тысячам см–1. Причем полосы поглощения для одной и той же молекулы по разному сдвигаются в зависимости от растворителя. Например, спектр поглощения для молекулы 3-аминофталимида смещается в красную сторону в н-гептане на 1400 см–1, а в этиловом спирте и воде – до 2300 см–1.
В настоящее время установлены многие закономерности проявления межмолекулярных взаимодействий, имеющих самую разнообразную природу. Создан даже отдельный отдел спектроскопии межмолекулярных взаимодействий, который интенсивно развивается санкт-петербургской школой физиков (Бахшиев Н.Г., Либов В.С. и др.).
Источником информации в спектроскопии межмолекулярных взаимодействий о свойствах отдельных молекул служит не спектр молекулы как таковой, а изменение спектра при помещении молекулы в конденсированную среду.