

3.4. Определение энергии диссоциации двухатомной молекулы
3.4.1. Аналитический метод
Рассмотрим два метода определения диссоциации молекулы – аналитический и графический. При аналитическом способе нахождения энергии диссоциации необходимо анализировать двучленную формулу колебательной энергии:
![]()
и попытаться экстраполировать ее к границе диссоциации, соответствующей схождению колебательных уровней. Граница диссоциации наступит в том случае и при таких , когда разность энергий между соседними уровнями будет равна 0. Как видно в (3.3), колебательные интервалы линейно зависят от (если не учитывать вторые ангармоничности) и стремятся к нулю при приближении к своему максимальному значению maх. Возьмем разность между соседними колебательными уровнями у границы диссоциации и приравняем ее к нулю. Из полученного соотношения найдем maх. Итак,
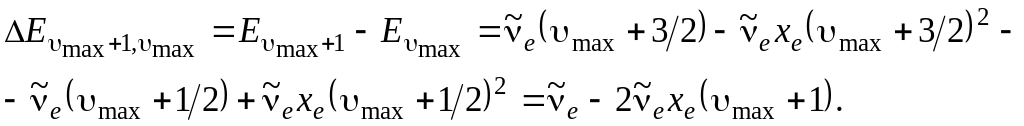 (3.41)
(3.41)
Приравняв это выражение нулю, получим 1 – 2хe (max + 1) = 0, откуда max = 1/2 хe ( учитывается тот факт, что хe << 1).
Подставляя max в выражение для колебательной энергии, получим значение энергии диссоциации молекулы:
![]() .
(3.42)
.
(3.42)
Необходимо отметить, что полученная формула весьма приближенная и дает несколько завышенные значения (примерно 17 %) энергии диссоциации. Формулу (3.42) легко получить и несколько иначе.
У границы диссоциации существует max, после которого следующий дискретный уровень с max + 1 будет отсутствовать. Граница диссоциации получается при таком значении , при котором E как функция достигает максимума. Если построить график зависимости E от (рис. 3.11), то функция E будет нелинейной и у границы диссоциации при max + 1 она должна была бы повернуть вниз, если бы такое max + 1 существовало (за счет существенного влияния квадратичного члена).
Максимум функции E соответствует условию
![]() .
(3.43)
.
(3.43)
Из последнего равенства получим
![]() .
(3.44)
.
(3.44)
Подстановка гран в формулу ( 3.42) дает значение De
![]() .
(3.45)
.
(3.45)

Расчетные значения энергии диссоциации не совпадают с экспериментально полученными значениями D0, которые меньше на величину нулевой энергии молекулы. Действительно, как видно из рисунка 3.7,
![]() ,
,
так как все внутренние значения энергии уничтожаются. Так как
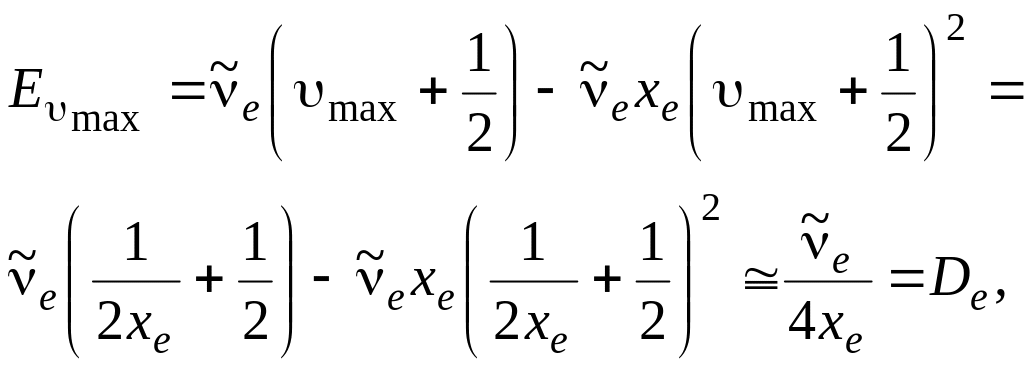
то
![]() .
(3.46 )
.
(3.46 )
Непосредственно
из опыта определяется D0,
а значение
находится
по формуле (3.45), если известны
![]() и хe
.
и хe
.
Если энергию отсчитывать от = 0, а не от минимума кривой потенциальной энергии, то двучленную формулу (3.29) можно записать в виде:
![]() .
(3.47)
.
(3.47)
Если
положить в этой формуле,
![]() а
а
![]() то равенство (3.47) можно переписать
следующим образом:
то равенство (3.47) можно переписать
следующим образом:
![]() ,
(3.48)
,
(3.48)
где
частота
![]() и постоянная ангармоничности х0
соответствуют уровню
= 0.
и постоянная ангармоничности х0
соответствуют уровню
= 0.
Энергию диссоциации D0 находим так, как и раньше. Определим max, при котором функция E – E0 максимальна. Взяв производную по от E – E0 и приравняв ее нулю, получим
![]() ,
,
откуда
![]() .
.
Подставив полученное значение в (3.48), получим
![]() .
( 3.49)
.
( 3.49)
Частоты переходов между соседними уровнями энергии на основании равенства (3.48) определим по формуле:
![]() .
(3.50)
.
(3.50)
Сумма первых разностей по всем колебательным квантовым числам до max, равна энергии диссоциации D0. Введение коэффициента + 1/2, а не в первые разности связано с определением энергии диссоциации.