

6.9. Электронные спектры молекул в твердотельных матрицах. Эффект Шпольского
Как уже было отмечено в п. 6.5, спектры поглощения и люминесценции красителей в растворах при комнатной температуре состоят обычно из одной или нескольких широких полос, расположенных в видимой и близкой ультрафиолетовой области. Широкие полосы объяснялись сравнительно быстрым колебательным перераспределением поглощенной энергии по колебательным подуровням и взаимодействием растворенной молекулы с окружающей средой. Ароматические молекулы в растворах характеризуются полосатыми спектрами (четыре – пять полос) с шириной полосы несколько сот обратных см–1 .
Полосы несколько сужаются при понижении температуры и даже в хорошо стеклующихся растворителях (смеси различных спиртов) ширина полос достигает сотни см–1 .
Более структурные спектры можно получить для ароматических кристаллов (бензол, нафталин, антрацен) при низких температурах порядка 4 К для образцов толщиной несколько мкм.. Однако, как оказалось несколько позже благодаря исследованиям Шпольского Э.В.и сотрудников, можно получить тонкоструктурные спектры органических молекул, не прибегая к выращиванию кристаллов. Если в качестве растворителей применить нейтральные, легко кристаллизующиеся нормальные парафины общей формулой Сh Нh+2 (пентан, гексан, гептан и др.), то для данного растворенного вещества (например, бензол, нафталин, антрацен) можно подобрать такой растворитель из класса нормальных парафинов, что в получающихся при замораживании твердых растворах спектры поглощения (аналогично и спектры люминесценции) имеют вид довольно узких линий 2–3 см–1, сгруппированных в «мультиплеты», которые зависят от рода растворителя и скорости охлаждения раствора. «Мультиплетами» их назвали по аналогии с линиями в атомных спектрах. Этот эффект в литературе получил название эффекта Шпольского и был открыт в 1952 г. Несколько позже спектры, в которых обнаруживается большое число узких полос (иногда более сотни), назвали квазилинейчатыми.
Рассмотрим происхождение квазилинейчатых спектров в замороженных кристаллических растворах. Для этого проведем сравнение спектров растворенных веществ в газовом и кристаллическом состояниях. Полосатая структура спектров молекул в газовой фазе объясняется вращательным уширением полос. При вращении молекулы уровни моменты инерции сравнительно велики, что приводит к малым значениям вращательных частот, которые будут группироваться вблизи электронно-колебательной линии и приводить к ее уширению. Разрешить вращательную структуру даже с помощью приборов высокого разрешения практически невозможно. В растворах, где вращение молекулы несколько заторможено, электронно-колебательный спектр уширен за счет релаксационно-ориентационных взаимодействий. В чистых кристаллах электронно-колебательные спектры состоят из довольно широких полос благодаря сильным электрон-фононным взаимодействиям, которые приводят к уширению уровней энергии. Как бы сильно ни уширяло уровни энергии межмолекулярное взаимодействие, в активированных кристаллах можно добиться такого состояния между поглощающими и излучающими молекулами, чтобы расстояние между ними было достаточно велико (малая концентрация молекул), а взаимодействие внедренной молекулы и матрицы было достаточно мало. Оказалось, что последний случай можно осуществить, если поглощающую или излучающую молекулу внедрить в кристаллическую решетку (матрицу), которая должна удовлетворять некоторым требованиям. Во-первых, внедренная молекула должна слабо взаимодействовать с кристаллом-основанием (матрицей). В этом случае кристалл должен состоять из молекул с насыщенными химическими связями, а атомы в молекуле не должны иметь неподеленных пар электронов, а сами внедренные молекулы не должны обладать в нижнем электронном состоянии дипольным моментом или во всяком случае их дипольные моменты должны быть достаточно малыми. Ароматические молекулы хорошо подходят во втором случае. Во-вторых, внедренная молекула не должна сильно деформироваться в кристалле-матрице, т.е. она должна хорошо располагаться (внедряться), почти изоморфно замещая молекулу основания или удобно встраиваться между узлами кристалла. Но при этом она не должна свободно вращаться, а фиксироваться в определенном положении, представляя собой некую упорядоченную систему, подобную ориентированному газу. В таких условиях молекула сохраняет электронные уровни энергий почти такими же, как и в парах, чего нельзя сказать о кристаллах. Осуществить указанные условия можно путем изоморфного замещения молекул кристалла-основания молекулами примеси. Во всех случаях, где возможно это было выполнить, молекулы дают квазилинейчатый спектр. Однако в случае органических соединений образование квазилинейчатых спектров возможно и при соблюдении более широких условий. Например, внедренная молекула должна хорошо расположиться между слоями молекул кристалла-основания. Рассмотрим это на примере нормальных парафинов, молекулы которых образуют неразветвленные линейные цепочки, например н-пентан, н-гексан. Соединения этого ряда различаются на одну группу СН2 и представляют собой большой набор молекул с аналогичными свойствами, но различной длины. Эти вещества легко кристаллизуются, прозрачны в видимой и ультрафиолетовой областях спектра и являются в химическом отношении несколько инертными (не реакционно-способными). Они принадлежат к другому классу, чем ароматические соединения, но имеют много подобных черт в своем строении.
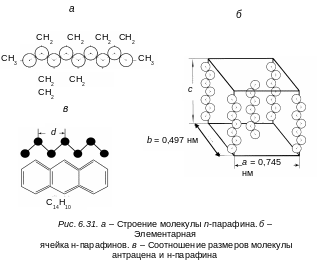
Нормальные
парафины представляют собой линейные
цепочки (см. рис. 6.31, а)
атомов углерода с расстоянием между
ними, равным 0,1–0,154
нм (см. рис. 6.31, а).
Рентгеноструктурное исследование
парафинов обнаружило, что элементарная
ячейка в направлениях осей
![]() и
и
![]() во всех парафинах имеет одинаковую
величину, а
= 0,745, b
=
0,497
нм. Размеры ячейки в направлении оси
во всех парафинах имеет одинаковую
величину, а
= 0,745, b
=
0,497
нм. Размеры ячейки в направлении оси
![]() линейно возрастают с числом атомов
углерода в молекуле парафина (см. рис.
6.31,
б).
Атомы углерода в цепочке парафина
расположены зигзагообразно. Наиболее
простые наглядные соотношения между
размерами молекулы активатора и матрицы
имеются для ароматических углеводородов
(нафталина, антрацена, нафтацена и др.).
Оказывается, что близость размеров
молекулы активатора и матрицы для этих
систем хорошо удовлетворяется (см. рис.
6.31,
в).
В тех случаях, когда получается хороший
квазилинейчатый спектр, имеется хорошее
соответствие меду размерами длинных
осей ароматической молекулы и того
парафина, который служит растворителем.
Только в этом случае получается резкий
квазилинейчатый спектр молекулы
ароматического соединения. В качестве
примера на рис. 6.22 приведен спектр
поглощения и флуоресценции нафталина
в н-пентане.
Как видно из рис.6.
22, мы
наблюдаем отдельные линии (квазилинии),
соответствующие переходам на колебательные
подуровни верхнего и нижнего электронных
состояний молекулы. Внутренние частоты
колебаний молекулы нафталина практически
не отличаются от аналогичных частот в
парах. Только частота
перехода несколько смещена в красную
сторону относительно той же частоты в
парах (см. табл.
6.2).
Такой же спектр флуоресценции нафталина
в гексане и гептане ухудшается по
сравнению со спектром в н-пентане
из-за большей свободы расположения
молекулы в кристаллической решетке.
Если же размеры молекулы матрицы меньше
размеров молекулы активатора, то
ухудшение спектра связано с деформацией
молекулы. Таким образом, когда имеется
хорошая квазилинейчатая структура
спектра, то между молекулами активатора
и матрицы существует не только
приблизительное равенство их длин, но
и геометрическое подобие зигзагов (см.
рис. 6.31, в).
линейно возрастают с числом атомов
углерода в молекуле парафина (см. рис.
6.31,
б).
Атомы углерода в цепочке парафина
расположены зигзагообразно. Наиболее
простые наглядные соотношения между
размерами молекулы активатора и матрицы
имеются для ароматических углеводородов
(нафталина, антрацена, нафтацена и др.).
Оказывается, что близость размеров
молекулы активатора и матрицы для этих
систем хорошо удовлетворяется (см. рис.
6.31,
в).
В тех случаях, когда получается хороший
квазилинейчатый спектр, имеется хорошее
соответствие меду размерами длинных
осей ароматической молекулы и того
парафина, который служит растворителем.
Только в этом случае получается резкий
квазилинейчатый спектр молекулы
ароматического соединения. В качестве
примера на рис. 6.22 приведен спектр
поглощения и флуоресценции нафталина
в н-пентане.
Как видно из рис.6.
22, мы
наблюдаем отдельные линии (квазилинии),
соответствующие переходам на колебательные
подуровни верхнего и нижнего электронных
состояний молекулы. Внутренние частоты
колебаний молекулы нафталина практически
не отличаются от аналогичных частот в
парах. Только частота
перехода несколько смещена в красную
сторону относительно той же частоты в
парах (см. табл.
6.2).
Такой же спектр флуоресценции нафталина
в гексане и гептане ухудшается по
сравнению со спектром в н-пентане
из-за большей свободы расположения
молекулы в кристаллической решетке.
Если же размеры молекулы матрицы меньше
размеров молекулы активатора, то
ухудшение спектра связано с деформацией
молекулы. Таким образом, когда имеется
хорошая квазилинейчатая структура
спектра, то между молекулами активатора
и матрицы существует не только
приблизительное равенство их длин, но
и геометрическое подобие зигзагов (см.
рис. 6.31, в).
Кристаллы н-парафинов имеют слоистый характер, что позволяет некоторым большим плоским молекулам располагаться между слоями. В большинстве случаев связь молекулы с решеткой кристалла настолько мала, что внедренные молекулы можно рассматривать как ориентированный газ, в спектр которого матрица не вносит существенных изменений, если не считать небольшого смещения частоты чисто электронного перехода. Однако в большинстве случаев возникают осложнения, которые заключаются в том, что вблизи линии 0–0 перехода возникает не одна, а целая группа (три-четыре) линий, резонансно совпадающих с соответствующими линиями в спектре поглощения при той же температуре. Возникает, как говорят спектроскописты, мультиплет, который затем повторяется в спектре с частотой полносимметричных колебаний (несущая частота). Эти группы линий условно назвали мультиплетами по аналогии с группами линий атомных спектров.
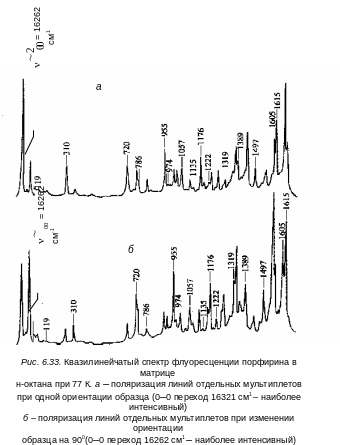
На рис. 6.32 приведены спектры поглощения и флуоресценции антрацена в н-гептане при 4,2 К. В спектрах поглощения все линии головного мультиплета резонансно совпадают с линиями того же «мультиплета» в спектре люминесценции.

Объяснить появление мультиплетов (три линии «мультиплетов») резонансными взаимодействиями растворенных молекул нельзя в силу низкой их концентрации.
Рассмотрим основные причины, приводящие к образованию мультиплетной структуры спектров поглощения и флуоресценции. Согласно одной из них мультиплетность квазилинейчатых спектров обусловлена существованием нескольких типов пространственно изолированных молекулярных центров, которые появляются при различной скорости кристаллизации образцов. Под влиянием различных локальных окружений, которые являются возмущением для электронной энергии примесных молекул, происходит смещение их электронных уровней. Наложение на чисто электронные переходы внутримолекулярных колебаний приводит к повторению «мультиплетов» по всему спектру. Изучение распределения интенсивности линий в компонентах «мультиплета» в зависимости от скорости замораживания образцов показали, что при более медленном охлаждении образуются более устойчивые центры свечения и некоторые слабые линии в спектрах могут исчезнуть совсем.
Вторым убедительным подтверждением принадлежности компонентов «мультиплетов» разным пространственно разделенным центрам явились исследования спектров возбуждения люминесценции ароматических молекул в матрицах н-алканов, выполненных при низких температурах учениками К. К. Ребане, Э. В. Шпольского и К. Н.Соловьева. Селективное возбуждение одной их компонент «мультиплета» приводит к исчезновению в спектре люминесценции компонент, принадлежащих молекулам других примесных центров. Это свидетельствует в пользу того, что отдельные компоненты «мультиплета» принадлежат различным центрам свечения.
Существует точка зрения, что происхождение «мультиплетов» в квазилинейчатых спектрах объясняется наличием нескольких поворотных изомеров в матрице н-алканов. В жидкости н-алканы представляют собой смесь поворотных изомеров. При быстрой кристаллизации молекулы различных изомеров не успевают принять устойчивую плоскую форму и могут включаться в решетку в виде поворотных изомеров, что и обусловливает многообразие кристаллических дефектов. Примесные молекулы, которые располагаются вблизи различных кристаллических дефектов, испытывают различное возмущение, что и приводит к появлению сложных по своей структуре «мультиплетов» в квазилинейчатых спектрах. При медленном охлаждении поворотные изомеры молекул н-алкана переходят в наиболее устойчивую плоскую форму, и «мультиплетная» структура квазилинейчатого спектра упрощается, число квазилиний уменьшается. Эта точка зрения также должна приниматься во внимание.
Следует отметить и еще одну существенную причину происхождения «мультиплетной » структуры квазилинейчатых спектров – это наличие нескольких кристаллических модификаций н-алкана в обычно используемых на опыте кристаллических образцах. Быстрое охлаждение может привести к тому, что в поликристалле образуются различные кристаллические формы, а примесные молекулы или ионы, попав в различные кристаллические ячейки, будут испытывать различное влияние кристаллического поля, что приведет к различному сдвигу их чисто электронных переходов (0–0 переходов).
В последнее время появились условия для надежного воспроизведения локальных кристаллических полей, в которых находится примесная молекула, путем получения хороших монокристаллов н-алканов и проведения поляризационных измерений для отдельных линий «мультиплета». Так, К. Н. Соловьев и его ученики предложили новый метод определения симметрии нормальных колебаний многоатомных молекул, внедренных в качестве примеси в монокристаллы н-алканов. Суть метода сводится к исследованию интенсивности отдельных квазилиний в зависимости от ориентации образца. Например, пусть имеется два типа центров флуоресценции, приводящих к дублетной структуре «мультиплета» квазилинейчатого спектра. Тогда поворотом монокристалла находятся два таких положения, в одном из которых интенсивность 0-0 перехода в спектре флуоресценции максимальна, а для молекул второго типа центров – минимальна. Минимальная интенсивность квазилиний соответствует тому случаю, когда ориентация излучающего диполя близка к направлению наблюдения флуоресценции. Квазилинии, соответствующие отдельным вибронным переходам в полученном по такой методике спектре флуоресценции, можно классифицировать по изменению интенсивности в зависимости от ориентации образца на две группы: интенсивность квазилиний одной группы изменяется так же, как интенсивность головной квазилинии (0–0 перехода), а второй группы ‑ противоположным образом . В первом случае активное колебание является полносимметричным (оно ответственно за появление серии линий и имеет поляризацию совпадающую с поляризацией чисто электронного перехода), а во втором – неполносимметричным. В последнем случае его поляризация не совпадает с поляризацией одной из компонент головного «мультиплета» (см. рис. 6.33). Аналогичным образом можно классифицировать по типам симметрии колебания, активные в спектре поглощения. Для этого необходимо исследовать интенсивность квазилиний в спектре возбуждения флуоресценции при регистрации интенсивности квазилинии 0‑0 перехода определенного типа центров в зависимости от ориентации активированного монокристалла. Наибольшие различия в интенсивности квазилиний для полносимметричных и неполносимметричных колебаний будет наблюдаться в тех же положениях образца, но относительно возбуждающего луча. Эти исследования квазилинейчатых спектров ароматических молекул в монокристаллах показали, что основной причиной «мультиплетности» линий в матрицах является различная ориентация молекул примеси в решетке кристалла.
Рассматривая теорию квазилинейчатых спектров Шпольского, Ребане и Хижняков обратили внимание на аналогию между механизмом возникновения в кристалле -линии с естественной шириной в эффекте Мессбауэра и узких «линий» в квазилинейчатом спектре. Эффект Мессбауэра заключается в испускании - квантов ядром атома. При этом часть энергии квантового перехода затрачивается на сообщение излучающему ядру кинетической энергии отдачи, которая затем сопровождается рождением фононов в решетке. Однако, Мессбауэр показал, что существует конечная вероятность и таких переходов, которые не сопровождаются рождением фононов в кристалле (бесфононные переходы). При этих переходах энергия излучаемого -кванта в точности равна энергии квантового перехода в ядре, и спектр этого излучения состоит из чрезвычайно узкой линии с естественной шириной. Все это возможно, если рассматривать проблему с квантовой точкой зрения, а не с классической. Существование нулевых колебаний в кристалле создает предел для передачи энергии решетке. Итак, испускание широкой полосы и учет узкой резонансной линии в эффекте Мессбауэра характеризует два независимых процесса, осуществляемых со своей вероятностью. В этом и состоит разгадка отсутствия влияния колебаний решетки на ширину линии без отдачи.
В механизме испускания квазилинейчатого спектра возникает та же картина, что и в эффекте Мессбауэра. Возникновение в кристалле чисто резонасных линий с шириной несколько обратных сантиметров вместо сотни см–1 , резонансный характер головных линий (0–0 переход) спектра – это те же проблемы, что и в эффекте Мессбауэра. Если молекула находится в кристалле, то влияние кристаллических колебаний должно сказываться на 0-0 переходе и на ширине линий. При флуоресценции должны неизбежно возникать тепловые потери, обуславливающие стоксовский сдвиг. Эти потери должны были бы расстроить резонанс между спектром флуоресценции и поглощения примерно так, как импульс отдачи расстраивает резонанс в случае -излучения. Однако для линий 0–0 переходов этого не происходит. Линии здесь совпадают в поглощении и флуоресценции с точностью до ошибки измерения (0,1 – 1 см–1 ). Источником размывания линий является рождение фононов решетки, сопровождающее электронный переход в примесной молекуле. Бесфононные линии могут возникнуть только при особых переходах, которые должны быть бесфононными, в полной аналогии с мессбауэровскими переходами «без отдачи».
Ребане и Хижняков показали, что при осцилляторном моделировании поглощения и излучения могут быть такие комбинации осцилляторов, при которых внутреннее состояние кристалла не изменяется. Это переходы между состояниями осциллятров с одинаковыми колебательными квантовыми числами, например, 0–0, 1–1, 2–2, и т.д. Поскольку при этих переходах внутреннее состояние кристалла не изменяется, то они ведут к возникновению узкой бесфононной линии. Так как молекулы примеси находятся в слегка различных условиях, то это будет приводить к небольшому неоднородному уширению линии, т.е. бесфононные линии на самом деле должны быть квазилиниями. При всех остальных переходах, когда квантовые числа осцилляторов начального и конечного состояний изменяются должен возникнуть размытый спектр, состоящий из бесфононных линий и размытого фона, аналогично тому, как это имеет место в эффекте Мессбауэра. Интенсивность полосы, включающей бесфононную линию и фононное крыло, определяется фактором Дебая – Валлера:
![]() ,
,
где I0 (T) ‑ интегральная интенсивность бесфононной линии, I (Т) ‑ интегральная интенсивность фононного крыла. Фактор Дебая – Валлера был введен в теории рассеяния нейтронов и рентгеновских лучей кристалла, где этим коэффициентом определялась вероятность упругого рассеяния.
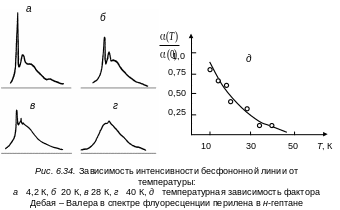
В нашем случае этот фактор определяет вероятность электронного перехода в примеси без изменения колебательного состояния кристаллической решетки. С другой стороны, фактор Дебая – Валлера может служить мерой силы электрон-фононного взаимодействия. Чем сильнее взаимодействие, тем меньше (Т). Повышение температуры приведет к монотонному убыванию (Т) (см. рис. 6.34).
При этом интегральная интенсивность всей полосы, включающей как бесфононную линию, так и ее фононное крыло, не изменяется. Однако при этом наблюдается падение интегральной интенсивности бесфононной линии, ее уширение и сдвиг и усиление фононного крыла с повышением температуры (см. рис. 6.34, а, б, в ,г).
Таким образом, теория примесных центров в кристаллах предсказывает существование в электронно-колебательных спектрах примеси узких бесфононных линий и широких полос, называемых фононными крыльями, отвечающих переходам в примеси с рождением и уничтожением фононов матрицы. Малая ширина линий казилинейчатых спектров и большое число их, а также высокая чувствительность положения линий к условиям окружения молекулы-активатора, делает эти спектры источником богатой информации о процессах, происходящих внутри молекулы и самого кристалла матрицы.
Благодаря развитию методов тонкоструктурной спектроскопии, особенно с применением лазеров, возникло новое перспективное направление в электронной спектроскопии – тонкоструктурная селективная спектроскопия, которая с успехом применяется для изучения спектров в матрицах Шпольского и свободных молекул, охлажденных в газовых потоках.