

Вращательные постоянные и структурные параметры некоторых
пирамидальных четырехатомных молекул
-
Молекула
В, см–1
r, нм
(град)
NH3
9,940
0.1014
106° 47'
NF3
0,3563
0,1371
102° 9'
РН3
4.4522
0,1421
93°27'
PF3
0,2608
0,1550
102°
PCl335
0,0874
0.2043
100°б'
Необходимо заметить, что при всех определениях структурных параметров молекул не учитываются нулевые колебания, присутствующие в молекуле. При определении масс атомов из спектроскопических данных наличие нулевых колебаний вызовет ошибку в определении массы приблизительно того же порядка, что и для линейных молекул.
Конечно, знание спектров КР и ИК-поглощения молекул в газовой фазе позволяет определять не только геометрические параметры молекулы, но и ряд других параметров центробежного растяжения, а по интенсивности и ширине соответствующих линий можно определять компоненты тензора поляризуемости, доплеровское уширение, сечение рассеяния и др.
Развитие и применение СКР тесно связано с развитием источников возбуждающего излучения высокой яркости и мощности (лазерных источников возбуждения). Еще большие возможности открывает новый нелинейно-оптический метод спектроскопии комбинационного рассеяния, известный под названием КАРС (спектроскопия когерентного антистоксового рассеяния света). Этот метод позволяет получить более высокий уровень сигналов (примерно в 105 раз выше, чем в СКР) и улучшить разрешение линий в спектрах до 0,001 см–1. Разрешение в методе СКР порядка 0,06 см–1.
Глава 3.
КОЛЕБАТЕЛЬНО-ВРАЩАТЕЛЬНЫЕ СПЕКТРЫ ДВУХАТОМНЫХ МОЛЕКУЛ
3.1.Образование двухатомной молекулы. Представление о ее потенциальной кривой.
В химически устойчивой двухатомной молекуле между атомами действуют силы преимущественно электрического притяжения. Величина и характер этих сил зависят от перераспределения электронной плотности внешних (валентных) электронов. Рассмотрим двухатомную молекулу из образующие ее атомы с точки зрения зависимости их потенциальной энергии от расстояния между ними.
П ока
атомы находятся на достаточно большом
расстоянии друг от друга, сила их
взаимодействия очень мала. Она появляется
в результате притяжения, обусловленного
взаимной поляризацией электронных
оболочек атомов. Затем сила притяжения
возрастает при уменьшении расстояния
между атомами, однако это возрастание
не беспредельно. на
малых сравнительно расстояниях между
атомами в действие вступают силы
отталкивания, обусловленные взаимным
влиянием ядер, а также проникновением
электронных оболочек, что и проявляется
в специфическом отталкивании между
электронами. Потенциальная энергия
отталкивания изображена на рис. 3.1
(кривая 1).
ока
атомы находятся на достаточно большом
расстоянии друг от друга, сила их
взаимодействия очень мала. Она появляется
в результате притяжения, обусловленного
взаимной поляризацией электронных
оболочек атомов. Затем сила притяжения
возрастает при уменьшении расстояния
между атомами, однако это возрастание
не беспредельно. на
малых сравнительно расстояниях между
атомами в действие вступают силы
отталкивания, обусловленные взаимным
влиянием ядер, а также проникновением
электронных оболочек, что и проявляется
в специфическом отталкивании между
электронами. Потенциальная энергия
отталкивания изображена на рис. 3.1
(кривая 1).
с уменьшением расстояния она возрастает быстрее, нежели энергия притяжения (кривая 2). Благодаря одновременному действию двух сил противоположного направления (сил притяжения и отталкивания) на некотором расстоянии между атомами установится равновесие: силы отталкивания равны силам притяжения. Потенциальная энергия системы, понимаемая как результирующая сумма энергий притяжения и отталкивания (рис. 3.2), будет иметь минимум. Расстояние между ядрами, которое соответствует минимуму потенциальной энергии, будем называть равновесным и обозначать .
При расстоянии между ядрами меньшем равновесного, превалируют силы отталкивания, поэтому потенциальная энергия будет неограниченно и быстро возрастать (левая ветвь потенциальной кривой на рис. 3.2). При увеличении расстояния между ядрами атомов, когда r → , потенциальная энергия должна расти до нуля, так как свободные атомы, удаленные на бесконечно большое расстояние друг от друга, не взаимодействуют и потенциальной энергией не обладают (правая ветвь потенциальной кривой рис. 3.2).
П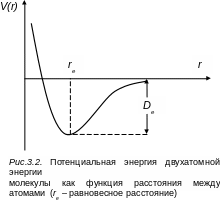 отенциальная
кривая V(r)
весьма характерна для химической связи
в двухатомной молекуле. Устойчивая
молекула будет существовать в том
случае, когда V(r)
имеет минимум. Значение r
будет
находиться в интервале 0 <
r
< .
Если минимум
отсутствует, то это означает, что при
сближении атомов не образуется устойчивой
молекулы. Первый случай соответствует
притяжению атомов при их сближении, а
второй –
отталкиванию.
Расстояние
отенциальная
кривая V(r)
весьма характерна для химической связи
в двухатомной молекуле. Устойчивая
молекула будет существовать в том
случае, когда V(r)
имеет минимум. Значение r
будет
находиться в интервале 0 <
r
< .
Если минимум
отсутствует, то это означает, что при
сближении атомов не образуется устойчивой
молекулы. Первый случай соответствует
притяжению атомов при их сближении, а
второй –
отталкиванию.
Расстояние
![]() ,
при котором энергия взаимодействия
имеет минимум, обычно принимают
за длину
химической связи.
,
при котором энергия взаимодействия
имеет минимум, обычно принимают
за длину
химической связи.
Расстояние
от минимума кривой до оси абсцисс, к
которой кривая асимптотически приближается
в своей правой части, соответствует
работе, которую необходимо затратить
для разрыва связи, а следовательно и
молекулы на составляющие ее атомы или
ионы и переноса их в бесконечность
(диссоциации). Энергия диссоциации
![]() = V
(r
=
)
–
V
(r
=
re)
(энергия
связи атомов в молекуле). Это определение
энергии не учитывает значения нулевой
колебательной энергии у молекулы,
поэтому определенная на опыте энергия
диссоциации будет меньше вычисленной.
= V
(r
=
)
–
V
(r
=
re)
(энергия
связи атомов в молекуле). Это определение
энергии не учитывает значения нулевой
колебательной энергии у молекулы,
поэтому определенная на опыте энергия
диссоциации будет меньше вычисленной.