

3.4.2. Метод графической экстраполяции (метод Берджа – Шпонер)
Сущность метода графической экстраполяции заключается в том, что строится график зависимости колебательных интервалов E + 1,, от + 1, который экстраполируется до пересечения с осью абсцисс. Площадь под этой кривой соответствует энергии диссоциации De. Заштрихованная область дает нижний предел . На рис. 3.12 приведен график этой зависимости для молекулы водорода.
При справедливости двучленной формулы для колебательной энергии (3.30), график является прямой линией и соответствующая площадь выражается формулой
![]() .
( 3.51)
.
( 3.51)

При
небольших отступлениях от линейности
получаются несколько иные значения
диссоциации. Действительно, при линейной
зависимости первой разности
![]() ордината в нулевой точке (
+
1),
т. е. при + 1=0,
равна
.
В точке, для которой ордината обращается
в нуль, значение абсциссы
ордината в нулевой точке (
+
1),
т. е. при + 1=0,
равна
.
В точке, для которой ордината обращается
в нуль, значение абсциссы
![]() ,
как это следует из (3.41). Площадь треугольника
определяется формулой
,
как это следует из (3.41). Площадь треугольника
определяется формулой
![]() ,
что согласуется с (3.42). Здесь энергию
диссоциации
отсчитывают от минимума потенциальной
энергии. Реальную же энергию диссоциации
D0
следует
отсчитывать от уровня
=
0,
для которого нулевая энергия равна
,
что согласуется с (3.42). Здесь энергию
диссоциации
отсчитывают от минимума потенциальной
энергии. Реальную же энергию диссоциации
D0
следует
отсчитывать от уровня
=
0,
для которого нулевая энергия равна
![]() ,
,
а энергия диссоциации
D0 = De – E0 . (3.52)
Частоты переходов между соседними уровнями, как следует из (3.50), равны
![]() ,
(3.53)
,
(3.53)
где
отличается от
на величину ангармоничности
![]() .
Следовательно
.
Следовательно
![]() ,
т. е. величина
практически совпадает с экспериментально
измеряемой частотой
,
т. е. величина
практически совпадает с экспериментально
измеряемой частотой
![]() .
( 3.54)
.
( 3.54)
При
графическом определении энергии
диссоциации D0
нужно
откладывать по оси абсцисс
+
1/2,
а по оси ординат
![]() .
Из формулы (3.50) получаем прежний график
(прямую линию), что на рис. 3.12 только
смещенный вправо на 1/2
(см. рис.
3.13).
.
Из формулы (3.50) получаем прежний график
(прямую линию), что на рис. 3.12 только
смещенный вправо на 1/2
(см. рис.
3.13).
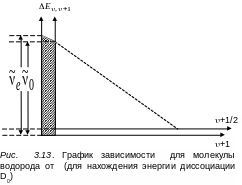
Разница
значений
и
D0
выражается
площадью, заштрихованной на рис. 3.13. Оси
абсцисс на рис. 3.13 совпадают. Действительно,
при
+
1,
согласно (3.35)
![]() а при
а при![]() согласно (3.50)
согласно (3.50)
![]() .
Заштрихованная площадь равна
.
Заштрихованная площадь равна
![]() (нулевая
энергия).
(нулевая
энергия).
Здесь
учтено, что
.
Следовательно, площадь треугольника
S
в координатах
и
![]() меньше площади другого треугольника
на величину E0.
Итак, D0
= De
–
E0
или же
меньше площади другого треугольника
на величину E0.
Итак, D0
= De
–
E0
или же
![]() .
.
П олучаемые
значения энергии диссоциации
и D0,
рассчитываемые описанными выше методами,
отличаются от истинных примерно на
15 %, что указывает на то, что формула
Морзе, аппроксимирующая реальную кривую
потенциальной энергии, является лишь
грубым приближением. Экспериментальные
точки ложатся не на прямую, а на кривую,
которая загибается раньше, т. е. пересекает
ось абсцисс при меньших значениях ,
чем это следует из анализа двучленной
формулы (3.30). На рис. 3.12 и 3.14 показаны
кривые зависимости E+1,
от квантового
числа
для молекул H2
и
Li2.
олучаемые
значения энергии диссоциации
и D0,
рассчитываемые описанными выше методами,
отличаются от истинных примерно на
15 %, что указывает на то, что формула
Морзе, аппроксимирующая реальную кривую
потенциальной энергии, является лишь
грубым приближением. Экспериментальные
точки ложатся не на прямую, а на кривую,
которая загибается раньше, т. е. пересекает
ось абсцисс при меньших значениях ,
чем это следует из анализа двучленной
формулы (3.30). На рис. 3.12 и 3.14 показаны
кривые зависимости E+1,
от квантового
числа
для молекул H2
и
Li2.

Энергия диссоциации определяется как площадь, ограниченная осями координат и реальной кривой, измеренная в соответствующем масштабе.
Энергию диссоциации можно измерить не только спектроскопическими, но и другими методами, которые мы здесь не рассматриваем (например, методом электронного удара, термическим или термохимическим. Знание энергий диссоциации молекул необходимо для термодинамических расчетов в химических реакциях.
Следует отметить, что задача определения энергии диссоциации по спектрам не проста, как может показаться из приведенного изложения. Требуется полная и точная расшифровка колебательного спектра, что не всегда можно довольно полно провести. Например, молекулы Н2, N2 и О2 при обычных условиях не поглощают ИК-радиации (у них отсутствует дипольный момент). Колебательные частоты у аналогичных молекул получают из спектров КР или из ИК-спектров при высоких давлениях, когда при соударениях симметрия (Dh) молекулы нарушается и могут наблюдаться оптически неактивные полосы.