

12. Титриметрія: розбіжність точок –стехіометрії та кінцевої
12.1. Загальні уявлення
Зміна загальних концентрацій у титруванні. Змішуючись, розчини розводять один одного. Спрощуючи розрахунок, вважають, що наближено об’єм суміші дорівнює сумі об’ємів розчинів, що складають цю суміш,
V = VA + VT, (12.1)
де, як у главі 11, індекс А відносимо до аналіту, а індекс Т – до титранту. Якщо концентрації аналіту та титранту відповідно дорівнюють с0(А) та с0(Т), то їх концентрації у суміші дорівнюють
c(A) = c0(A) VA / V, (12.2a)
c(T) = c0(T) VT / V, (12.2b)
де індекс 0 відноситься до вихідних, ще не змішаних, розчинів. У главі 11 цей індекс був непотрібний, бо взаємне розведення не відігравало ролі у стехіометричних розрахунках. У формулах (12.1) та (12.2) використовуємо співвідношення (11.3), що тут набуває вигляду
VT, st = {VA c0(A) / A } / {c0(T) / T}. (12.3)
Формули (12.1) та (12.2) не підставляємо у (12.3) – навіщо громіздкі перетворення, коли досить здійснити послідовні розрахунки!
Значна частина проблем теорії використовує рівноважний склад сумішей, близьких до точки стехіометрії. Спрощуючи розрахунки, наближено вважатимемо, що об’єм, знаменник у формулах (12.2), однаковий в усіх сумішах – такий, як у точці стехіометрії.
Похибка титрування. Титруючи, реєструють не точку стехіометрії, а “кінцеву точку”. Розбіжність між ними спричиняє похибку. В околі точки стехіометрії характерною є мала буферність, і відхили в доданку титранту значно впливають на рівноважні концентрації. Так легше помітити пов’язану з ними зміну властивостей. Кінцеву точку визначають, спостерігаючи залежність між витратою титранту та властивостями, такими як світлопоглинання, потенціал електроду, електропровідність розчину тощо. Простим є візуальне спостереження за зміною кольору розчину, до якого додано невелику кількість індикатору.
Методичну складову похибки, спричиненої розбіжністю між кінцевою точкою титрування та точкою стехіометрії, пов’язують із різними величинами. Абсолютна похибка у кількості речовини та об’ємі титранту – це
n(T) = n(T) fin ‑ n(T) st, (12.4a)
VT = VT, fin ‑ VT, st, (12.4b)
де індекс fin відповідає кінцевій точці титрування.
Відносна похибка дорівнює відношенню абсолютної похибки до величини, що вимірюють,
= n(T) / n(T) st = VT / VT, st. (12.5)
Крива титрування – графік залежності деякої властивості суміші від кількості уведеного в цю суміш титранту. Як властивість (ординату кривої) часто використовують показник активності (чи рівноважної концентрації) одного з учасників реакцій – величину рХ, яку часто можливо виміряти. На вісь абсцис можна наносити різні шкали (або декілька шкал), такі як n (T), n (fT T), VT. При малій буферності відносно Х в околі точки стехіометрії (розділи 5.2 та 5.5) уже малі доданки реагентів змінюють [X] на декілька порядків (“стрибок” у титруванні).
Будуючи теоретичну криву, розраховують рівноважний склад сумішей. Зручною абсцисою є частка доданого титранту від того, що відповідає точці стехіометрії, безрозмірна відносна величина
= VT / VT st = n(T) / n st(T) = 1 + . (12.6)
Індикатори. У пошуку кінцевої точки простим сигналом є зміна забарвлення доданого індикатору. Надмірна його концентрація шкодить, бо незручно спостерігати за забарвленням, коли поглинається значна частина світла. Сприйняття забарвлення залежить і від освітлення (денне світло чи вольфрамова лампа). У довідниках подають інтервали рХ = ‑ lg [X], що відповідають зміні забарвлення індикаторів й охоплюють границю переважання їх форм. Якщо відсутні довідкові дані, вважають, що на кінцях інтервалу співвідношення концентрацій форм індикатору 1 : 10. Для кислотно-основних індикаторів, що приєднують чи втрачають іон Н+, границі інтервалу – це наближено рН = lg КН 1.
Підвищуючи точність індикації, забарвлення порівнюють із таким для “свідка” – розчину, що за складом імітує точку стехіометрії із індикатором. Інший спосіб – вживати змішаний індикатор. Це або суміш двох різних індикаторів, інтервали переходу яких перетинаються, або суміш індикатору з барвником, що не змінює забарвлення. Корисні суміші, компоненти яких у кінцевій точці мають додаткові кольори, світло рівномірно поглинається у всій видимій області, й забарвлення стає слабко сірим. Їх інтервал pХ звужується до 0,2. Приклад суміші індикатору й барвника – метиловий оранжевий й індигокармін, що в лужній області жовто-зелена, у кислій фіолетова, а при pH 4 сіра.
Приклад 12.1. Крива титрування сильної кислоти сильною основою.
Розв’язок. Реакція (11.1) тут –
H+ + OH‑ H2O.
Координати (, pH) точок кривої титрування при заданих c0(H+) та c0(OH‑) обчислюємо, як у прикладі 5.1. Величина , за співвідношенням (12.6), задає методичну складову похибки. На рис. 12.1 подано криву для однакових концентрацій, c0(OH‑) = c0(H+) = 0,20 моль/л, й
Vst(OH‑) = V(H+), Vst = V(H+) + Vst(OH‑) = 2 V(H+),
отже, у точці стехіометрії c(OH‑)st = c(H+)st = 0,10 моль/л. Пунктиром зображено результати розрахунку в наближенні, що об’єм суміші розчинів, V, є постійним – таким, як у точці стехіометрії,
t(H+) {c0(H+) VH – c0(OH‑) VOH} / (2 VH) =
= c0(H+) / 2 – c0(OH‑) VOH / (2 VH).
Перед точкою стехіометрії майже всюди справедливе наближення [H+] << [OH‑], після неї [H+] >> [OH‑], а в її околі (при 1) лишаємо обидва доданки, зводячи рівняння до квадратного за [H+] = 10‑pH.
Якщо аналітом є основа, а титрантом – кислота, то рівняння кривої відрізняється знаком перед дробом у середині низки рівностей та взаємною заміною c0(H+) на c0(OH‑). Крива є не зростаючою, а спадною.
Задачі теорії титриметрії:
(1) оцінити методичну складову похибки (12.5), що відповідає методові фіксації кінцевої точки;
(2) вибрати метод фіксації кінцевої точки, що забезпечує задані вимоги до методичної складової похибки.
Першу з них вирішують, не розраховуючи усієї кривої титрування. За методами глави 8 можна визначити витрату титранту для рХfin, пов’язаного зі зміною забарвлення індикатору «показника титрування».
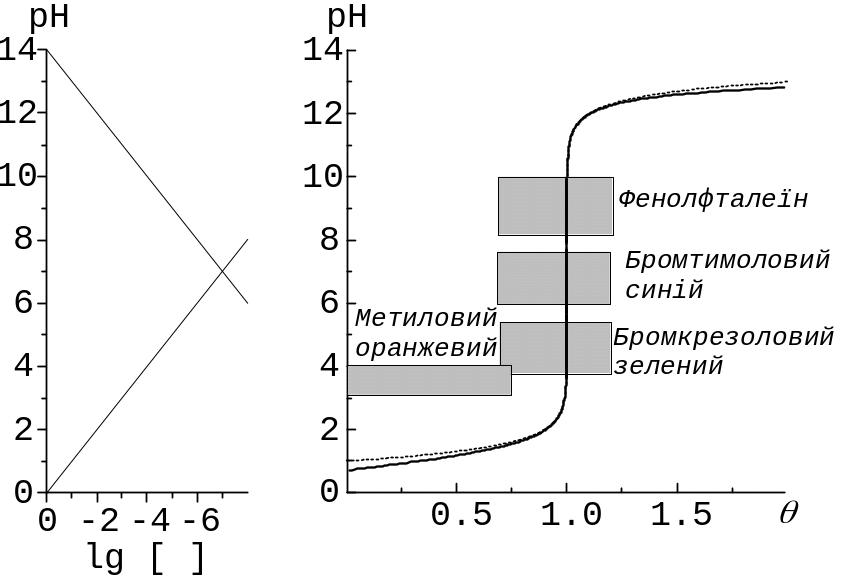
Рис. 12.1. Крива титрування сильної кислоти сильною основою при c0(H+) = c0(OH‑) = 0,20 моль/л. Пунктирний графік – у наближенні, де об’єм суміші розчинів є постійним, таким, як у точці стехіометрії. Ліворуч – КЛД з осями, що повернуті на 90o порівняно з традиційними (щоб сумістити осі pH). Заштриховано області зміни забарвлення індикаторів: метилового оранжевого (3,1 < pH < 4,0), бромфенолового зеленого (3,8 < pH < 5,4), бромтимолового синього (6,0 < pH 7,6), фенолфталеїну (8,2 < pH < 10,0).
Нехай у прикладі 12.1 рНfin = (3,1 + 4,0) / = 3,55 – на інтервалі переходу для метилового оранжевого. При = 1 маємо
[H+] = 10‑pH = 10‑3,55 = 2,810‑4 моль/л,
[OH‑] = ![]() = 10‑10,45 = 3,510‑11 моль/л,
[H+] ‑ [OH‑],
c(OH‑) = ‑ t(H+) = [OH‑] ‑ [H+] = ‑ 2,810‑4 моль/л.
= 10‑10,45 = 3,510‑11 моль/л,
[H+] ‑ [OH‑],
c(OH‑) = ‑ t(H+) = [OH‑] ‑ [H+] = ‑ 2,810‑4 моль/л.
Взаємне розведення розчинів дає c(OH‑)st = 0,10 моль/л, відносна похибка ‑
с(OH‑) / с(OH‑) st = ‑ 2,810‑4 / 0,10 = ‑ 2,810‑3 = ‑ 0,28 %.
Друга із задач – вибір умов індикації, якщо задано граничне значення похибки, наприклад, || 0,001. Величини с(Т) та – монотонні функції доданка титранту. На кінцях інтервалу ‑0,001 < < 0,001 у наближенні V = const прикладу 12.1 маємо t(H+) = 110‑4 моль/л та t(H+) = ‑ 110‑4 моль/л. Для першого з них нехтуємо у балансі доданком [OH‑],
[H+] = 10‑pH t(H+) = 110‑4 моль/л, pH ‑ lg t(H+) = 4,0.
Для другого із цих t(H+) нехтуємо доданком [H+], тоді
[OH‑] = ‑ t(H+) = 110‑4 моль/л,
pH + lg Kw lg {‑ t(H+)} = ‑ 4,0, pH = ‑ 4,0 + 14,0 = 10,0.
Знайдений інтервал 4,0 < рН < 10,0 на рис. 12.1 охоплює інтервали переходу для індикаторів бромтимолового синього (6,0 < pH < 7,6), фенолфталеїну (8,2 < pH < 10,0) і кінці інтервалів для метилового оранжевого (3,1 < pH < 4,0) та бромфенолового зеленого (3,8 < pH < 5,4).
Для теоретичних досліджень розроблено різні алгоритми. Їх автори часто спокушаються як аргумент у формулах похибок вживати похибку в рХ, спричинену індикацією,
pХ = рХfin ‑ рХst , (12.7)
і як крок 1 подають розрахунок рХst (наприклад, рНst). Типовою є формула для титрування слабкої основи сильною кислотою,
= (10 ‑ pH ‑ 10 pH) / {t(B) KH}1/2, (12.8)
де враховано найбільші рівноважні концентрації у рівнянні балансу. На кроці 2 за знаком pН визначають, нестачі чи надлишкові титранту відповідає кінцева точка. Нарешті за формулою (12.8) обчислюють .
Оцінюючи , стикаємось з проблемою похибки для малих pХ Теорію оцінок часто ускладнюють, поділяючи похибку на складові – систематичну та випадкову [11, с. 195]. У кислотно-основному титруванні середній квадратичний відхил рН прирівнюють 0,4 (за індикатором) та 0,2 (із застосуванням розчину “свідка”). Похибку перераховують на витрату титранту, множачи її на буферну ємність, та на 3 («правило 3 сiгма»). Загальна похибка – це, приблизно, більша із знайдених складових. На наш погляд, рХfin слід розглядати не як точне значення, а як інтервал. Випадкову складову окремо уводити не варто – досить узяти найбільшу із оцінок похибки на кінцях інтервалу. У формулах типу (12.8) обмежуємось більшим за абсолютною величиною доданком у чисельнику.
Більш наочно і просто в оцінках використовувати безпосередньо фрагменти КЛД. Якщо вжито приблизний ескіз цих фрагментів, то він допомагає швидко і легко вивести метод уточнення похибки розрахунком, що краще, ніж механічно і несвідомо застосовувати готові формули.