

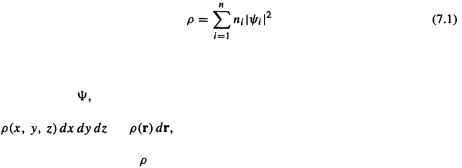
Density Functional Calculations 387
7.2THE BASIC PRINCIPLES OF DENSITY FUNCTIONAL THEORY
7.2.1 Preliminaries
In the Born interpretation (section 4.2.6) the square of a one-electronwavefunction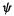 at any point X is the probability density (with units of
at any point X is the probability density (with units of  for the wavefunction at that point, and
for the wavefunction at that point, and  is the probability (a pure number) at any moment of finding the electron in an infinitesimal volume dx dy dz around the point (the probability of finding the electron at a mathematical point is zero). For a multielectron wavefunction
is the probability (a pure number) at any moment of finding the electron in an infinitesimal volume dx dy dz around the point (the probability of finding the electron at a mathematical point is zero). For a multielectron wavefunction  the relationship between thewavefunction
the relationship between thewavefunction 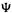 and the electron density
and the electron density  is more complicated (involving the summation over all spin states of all electrons of n-fold integrals of the square of the wavefunction), but it can be shown [7] that
is more complicated (involving the summation over all spin states of all electrons of n-fold integrals of the square of the wavefunction), but it can be shown [7] that  is related to the component one-electron spatialwavefunctions
is related to the component one-electron spatialwavefunctions  of a single-determinant wavefunction
of a single-determinant wavefunction (recall from section 5.2.3.1 that the Hartree–Fock
(recall from section 5.2.3.1 that the Hartree–Fock 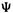 can be approximated as a Slater determinant of spin orbitals
can be approximated as a Slater determinant of spin orbitals  and
and  by
by
The sum is over n the occupied MOs 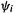 and for a closed-shell molecule each
and for a closed-shell molecule each 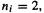 for a total of 2n electrons. Equation (7.1) applies strictly only to a single-determinant wavefunction but for multideterminant wavefunctions arising from configuration interaction treatments (section 5.4) there are similar equations [8]. A shorthand for
for a total of 2n electrons. Equation (7.1) applies strictly only to a single-determinant wavefunction but for multideterminant wavefunctions arising from configuration interaction treatments (section 5.4) there are similar equations [8]. A shorthand for
is |
where r is the position vector of the point with |
coordinates (x, y, z). |
|
If the electron density |
rather than the wavefunction could be used to calculate |
molecular geometries, energies, etc., this might be an improvement over the wavefunction approach because, as mentioned above, the electron density in an n -electron molecule is a function of only the three spatial coordinates x, y, z, but the wavefunction is a function of 4n coordinates. Density functional theory seeks to calculate all the properties of atoms and molecules from the electron density. The standard book on DFT is probably still that by Parr and Yang (1989) [9]; more recent developments are included in the book by Koch and Holthausen [10]. The textbook by Levine [11] gives a very good yet compact introduction to DFT, and among the many reviews are those by Friesner et al. [12], Kohn et al. [13], and Parr and Yang [14].
7.2.2 Forerunners to current DFT methods
The idea of calculating atomic and molecular properties from the electron density appears to have arisen from calculations made independently by Enrico Fermi and P. A. M. Dirac in the 1920s on an ideal electron gas, work now well-known as the Fermi–Dirac statistics [15]. In independent work by Fermi [16] and Thomas [17], atoms were modelled as systems with a positive potential (the nucleus) located in a uniform (homogeneous) electron gas. This obviously unrealistic idealization, the

 method, which was developed mainly for atoms and solids, has also been used for molecules, but has been replaced by the more accurate Kohn–Sham type (section 7.2.3) DFT methods.
method, which was developed mainly for atoms and solids, has also been used for molecules, but has been replaced by the more accurate Kohn–Sham type (section 7.2.3) DFT methods. In other words, given
In other words, given  we can in principle calculate any ground state property, e.g. the energy,
we can in principle calculate any ground state property, e.g. the energy, 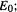 we could represent this as
we could represent this as is
is 
 transforms the function
transforms the function  into the number 4. We designate the fact that the integral is a functional of
into the number 4. We designate the fact that the integral is a functional of 
Density Functional Calculations 389
The theorem is “merely” an existence theorem: it says that a functional F exists, but does not tell us how to find it; this omission is the main problem with DFT. The significance of this theorem is that it assures us that there is a way to calculate molecular properties from the electron density, and that we can infer that approximate functionals will give at least approximate answers.
The second Hohenberg–Kohn theorem [24] is the DFT analogue of the wavefunction variation theorem that we saw in connection with the ab initio method (section 5.2.3.3): it says that any trial electron density function will give an energy higher than (or equal to, if it were exactly the true electron density function) the true ground state energy. In DFT molecular calculations the electronic energy from a trial electron density is the energy of the electrons moving under the potential of the atomic nuclei. This nuclear potential is called the “external potential” and designated  and the electronic energy is denoted by
and the electronic energy is denoted by  (“the
(“the  functional of the ground state electron density”). The second theorem can thus be stated
functional of the ground state electron density”). The second theorem can thus be stated
where  is a trial electronic density and
is a trial electronic density and  is the true ground state energy, corresponding to the true electronic density
is the true ground state energy, corresponding to the true electronic density  The trial density must satisfy the conditions
The trial density must satisfy the conditions  where n is the number of electrons in the molecule (this is analogous to the wavefunction normalization condition; here the number of electrons in all the infinitesimal volumes must sum to the total number in the molecule) and
where n is the number of electrons in the molecule (this is analogous to the wavefunction normalization condition; here the number of electrons in all the infinitesimal volumes must sum to the total number in the molecule) and  for all r (the number of electrons per unit volume cannot be negative). This theorem assures us that any value of the molecular energy we calculate from the Kohn–Sham equations (below, a set of equations analogous to the Hartree–Fock equations, obtained by minimizing energy with respect to electron density) will be greater than or equal to the true energy (this is actually true only if the functional used were exact; see below). The Hohenberg–Kohn theorems were originally proved only for nondegenerate ground states, but have been shown to be valid for degenerate ground states too [25]. The functional of the inequality (7.6) is the correct, exact energy functional (the prescription for transforming the ground state electron density function into the ground state energy). The exact functional is unknown, so actual DFT calculations use approximate functionals, and are thus not variational: they can give an energy below the true energy. Being variational is a nice characteristic of a method, because it assures us that any energy we calculate is an upper bound to the true energy. However, this is not an essential feature of a method: Møller–Plesset and practical configuration interaction calculations (sections 5.4.2, 5.4.3) are not variational, but this is not regarded as a serious problem.
for all r (the number of electrons per unit volume cannot be negative). This theorem assures us that any value of the molecular energy we calculate from the Kohn–Sham equations (below, a set of equations analogous to the Hartree–Fock equations, obtained by minimizing energy with respect to electron density) will be greater than or equal to the true energy (this is actually true only if the functional used were exact; see below). The Hohenberg–Kohn theorems were originally proved only for nondegenerate ground states, but have been shown to be valid for degenerate ground states too [25]. The functional of the inequality (7.6) is the correct, exact energy functional (the prescription for transforming the ground state electron density function into the ground state energy). The exact functional is unknown, so actual DFT calculations use approximate functionals, and are thus not variational: they can give an energy below the true energy. Being variational is a nice characteristic of a method, because it assures us that any energy we calculate is an upper bound to the true energy. However, this is not an essential feature of a method: Møller–Plesset and practical configuration interaction calculations (sections 5.4.2, 5.4.3) are not variational, but this is not regarded as a serious problem.
7.2.3.2 The Kohn–Sham energy and the KS equations
The first Kohn–Sham theorem tells us that it is worth looking for a way to calculate molecular properties from the electron density. The second theorem suggests that a variational approach might yield a way to calculate the energy and electron density (the electron density, in turn, could be used to calculate other properties). Recall that in wavefunction theory, the Hartree–Fock variational approach (section 5.2.3.4) led to the HF equations, which are used to calculate the energy and the wavefunction. An analogous variational approach led (1965) to the KS equations [26], the basis of current
390 Computational Chemistry
molecular DFT calculations. The two basic ideas behind the KS approach to DFT are:
(1) To express the molecular energy as a sum of terms, only one of which, a relatively small term, involves the unknown functional. Thus even moderately large errors in this term will not introduce large errors into the total energy. (2) To use an initial guess of the electron density  in the KS equations (analogous to the HF equations) to calculate an initial guess of the KS orbitals (below); this initial guess is then used to refine these orbitals, in a manner similar to that used in the HF SCF method. The final KS orbitals are used to calculate an electron density that in turn is used to calculate the energy.
in the KS equations (analogous to the HF equations) to calculate an initial guess of the KS orbitals (below); this initial guess is then used to refine these orbitals, in a manner similar to that used in the HF SCF method. The final KS orbitals are used to calculate an electron density that in turn is used to calculate the energy.
The Kohn–Sham energy
The strategy here is to regard the energy of our molecule as a deviation from an ideal energy, which latter can be calculated exactly; the relatively small discrepancy contains the unknown functional, whose approximation is our main problem. The ideal energy is that of an ideal system, a fictitious noninteracting reference system, defined as one in which the electrons do not interact and in which the ground state electron density  is exactly the same as in our real ground state system:
is exactly the same as in our real ground state system:  We are talking about the electronic energy of the molecule; the total internal “frozen-nuclei” energy can be found later by adding the internuclear repulsions, and the 0 K total internal energy by further adding the zero-point energy, just as in HF calculations (section 5.2.3.6e).
We are talking about the electronic energy of the molecule; the total internal “frozen-nuclei” energy can be found later by adding the internuclear repulsions, and the 0 K total internal energy by further adding the zero-point energy, just as in HF calculations (section 5.2.3.6e).
The ground state electronic energy of our real molecule is the sum of the electron kinetic energies, the nucleus–electron attraction potential energies, and the electron–electron repulsion potential energies (more precisely, the sum of the quantummechanical average values or expectation values, each denoted 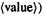 and each is a functional of the ground-state electron density:
and each is a functional of the ground-state electron density:
Focussing on the middle term: the nucleus–electron potential energy is the sum over all 2n electrons (as with our treatment of ab initio theory, we will work with a closedshell molecule which perforce has an even number of electrons) of the potential corresponding to attraction of an electron for all the nuclei A:
where  is the external potential (explained in section 7.2.3.1, in connection with Eq. (7.6)) for the attraction of electron i to the nuclei. The density function
is the external potential (explained in section 7.2.3.1, in connection with Eq. (7.6)) for the attraction of electron i to the nuclei. The density function  can be introduced into
can be introduced into  by using the fact [27] that
by using the fact [27] that
where  is a function of the coordinates of the 2n electrons of a system and
is a function of the coordinates of the 2n electrons of a system and 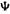 is the total wavefunction (the integrations are over spatial and spin coordinates on the left and spatial coordinates on the right). From Eqs (7.8) and (7.9), invoking the concept of
is the total wavefunction (the integrations are over spatial and spin coordinates on the left and spatial coordinates on the right). From Eqs (7.8) and (7.9), invoking the concept of

Density Functional Calculations 391
expectation value (chapter 5, section 5.2.3.3)  and since
and since we get
we get
So Eq. (7.7) can be written
Unfortunately, this equation for the energy cannot be used as it stands, since we do not know the functionals in and
and 
To utilize Eq. (7.11), Kohn and Sham introduced the idea of a reference system of noninteracting electrons. Let us define the quantity  as the deviation of the real kinetic energy from that of the reference system:
as the deviation of the real kinetic energy from that of the reference system:
Let us next define  as the deviation of the real electron–electron repulsion energy from a classical charge-cloud coulomb repulsion energy. This classical electrostatic repulsion energy is the summation of the repulsion energies for pairs of infinitesimal volume elements
as the deviation of the real electron–electron repulsion energy from a classical charge-cloud coulomb repulsion energy. This classical electrostatic repulsion energy is the summation of the repulsion energies for pairs of infinitesimal volume elements 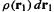 and
and  (in a nonquantum cloud of negative charge) separated by a distance
(in a nonquantum cloud of negative charge) separated by a distance multiplied by one-half (so that we do not count the
multiplied by one-half (so that we do not count the repulsion energy and again the
repulsion energy and again the  energy). The sum of infinitesimals is an integral and so
energy). The sum of infinitesimals is an integral and so
Actually, the classical charge-cloud repulsion is somewhat inappropriate for electrons in that smearing an electron (a particle) out into a cloud forces it to repel itself, as any two regions of the cloud interact repulsively. This physically incorrect electron self-interaction will be compensated for by a good exchange-correlation functional (below).
Using (7.12) and (7.13), Eq. (7.11) can be written as
The sum of the kinetic energy deviation from the reference system and the electron– electron repulsion energy deviation from the classical system is called the exchangecorrelation energy functional or the exchange-correlation energy,  (strictly speaking, the functional is the prescription for obtaining the energy from the electron density function):
(strictly speaking, the functional is the prescription for obtaining the energy from the electron density function):
The  term represents the kinetic correlation energy of the electrons and the
term represents the kinetic correlation energy of the electrons and the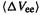 term the potential correlation energy and the exchange energy, although exchange and
term the potential correlation energy and the exchange energy, although exchange and

392 Computational Chemistry
correlation energy in DFT do not have exactly the same significance here as in HF theory [28];  is negative. So using Eq. (7.15), Eq. (7.14) becomes
is negative. So using Eq. (7.15), Eq. (7.14) becomes
Let us look at the four terms in the expression for the molecular energy  in Eq.(7.16).
in Eq.(7.16).
1. The first term (the integral of the density times the external potential) is
If we know the integrals to be summed are readily calculated.
the integrals to be summed are readily calculated.
2. The second term (the electronic kinetic energy of the noninteracting-electrons reference system) is the expectation value of the sum of the one-electron kinetic energy operators over the ground state multielectron wavefunction of the reference system (Parr and Yang explain this in detail [29]). Using the compact Dirac notation for integrals:
Since these hypothetical electrons are noninteracting 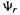 can be written exactly (for a closed-shell system) as a single Slater determinant of occupied spin molecular orbitals (section 5.2.3.1; for a real system, the electrons interact and using a single determinant causes errors due to neglect of electron correlation (section 5.4)). Thus, for a four-electron system,
can be written exactly (for a closed-shell system) as a single Slater determinant of occupied spin molecular orbitals (section 5.2.3.1; for a real system, the electrons interact and using a single determinant causes errors due to neglect of electron correlation (section 5.4)). Thus, for a four-electron system,
The 16 spin orbitals in this determinant are the KS spin orbitals of the reference system; each is the product of a KS spatial orbital  and a spin function
and a spin function  Equation (7.18) can be written in terms of the spatial KS orbitals by invoking a set of rules (the Slater– Condon rules [30]) for simplifying integrals involving Slater determinants:
Equation (7.18) can be written in terms of the spatial KS orbitals by invoking a set of rules (the Slater– Condon rules [30]) for simplifying integrals involving Slater determinants:
The integrals to be summed are readily calculated. Note that DFT per se does not involve wavefunctions, and the KS approach to DFT uses orbitals only as a way to calculate the noninteracting-system kinetic energy and the electron density function; see below.

Density Functional Calculations 393
3.The third term in Eq. (7.16), the classical electrostatic repulsion energy term, is readily calculated if 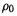 is known.
is known.
4.This leaves us with the exchange-correlation energy functional,  (Eq. (7.15)) as the only term for which some method of calculation must be devised.
(Eq. (7.15)) as the only term for which some method of calculation must be devised.
Devising accurate exchange-correlation functionals for calculating this energy term from the electron density function is the main problem in DFT research. This is discussed in section 7.2.3.4.
Written out more fully, then, Eq. (7.16) is
The term most subject to error is the relatively small  term, which contains the (exactly) unknown functional.
term, which contains the (exactly) unknown functional.
The Kohn–Sham equations
The KS equations are obtained by utilizing the variation principle, which the second Hohenberg–Kohn theorem assures us applies to DFT. We use the fact that the electron density of the reference system, which is the same as that of our real system (see the definition at the beginning of the discussion of the KS energy), is given by [7]
where the 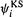 are the KS spatial orbitals. Substituting the above expression for the orbitals into the energy expression of Eq. (7.21) and varying
are the KS spatial orbitals. Substituting the above expression for the orbitals into the energy expression of Eq. (7.21) and varying with respect to the
with respect to the subject to the constraint that these remain orthonormal (the spin orbitals of a Slater determinant are orthonormal) leads to the KS equations (the derivation is given by Parr and Yang [31]; the procedure is similar to that used in deriving the Hartree–Fock equations, section 5.2.3.4):
subject to the constraint that these remain orthonormal (the spin orbitals of a Slater determinant are orthonormal) leads to the KS equations (the derivation is given by Parr and Yang [31]; the procedure is similar to that used in deriving the Hartree–Fock equations, section 5.2.3.4):
where |
are the KS energy levels (the KS orbitals and energy levels are discussed |
later) and |
(1) is the exchange correlation potential, arbitrarily designated here for |
electron number 1, since the KS equations are a set of one-electron equations (cf. the HF equations) with the subscript i running from 1 to n, over all the 2n electrons in the system. The exchange correlation potential is defined as the functional derivative of  with respect to
with respect to 