


Ab initio calculations 275
for CBS-Q, G2, G3, and G3(MP2) jobs on  OF were 59, 206, 136, and 41 minutes, respectively. It thus seems that where size permits and a slight loss of accuracy is tolerable, G3(MP2) is the method ofchoice. The maximum practical number ofheavy atoms for G2, CBS-Q, G2(MP2) and CBS-4 calculations were, at least recently, ca. 5, 7,7, and 15, respectively [124]. There are more accurate (and more time-consuming!) methods than any of the four in Table 5.10. The CBS-APNO method (available with a keyword in Gaussian 94 and 98), which is limited to about four heavy atoms [124], has a mean absolute deviation of only
OF were 59, 206, 136, and 41 minutes, respectively. It thus seems that where size permits and a slight loss of accuracy is tolerable, G3(MP2) is the method ofchoice. The maximum practical number ofheavy atoms for G2, CBS-Q, G2(MP2) and CBS-4 calculations were, at least recently, ca. 5, 7,7, and 15, respectively [124]. There are more accurate (and more time-consuming!) methods than any of the four in Table 5.10. The CBS-APNO method (available with a keyword in Gaussian 94 and 98), which is limited to about four heavy atoms [124], has a mean absolute deviation of only  [86], and a method that can give atomization energies accurate to about
[86], and a method that can give atomization energies accurate to about has been reported [125]. Because of the empirical correction terms, the Gaussian and CBS methods are not purely ab initio, except where these terms disappear by subtraction [123]. Some applications of high-accuracy energy methods and suggestions about choosing a method (high accuracy or otherwise) will be given in the following sections (5.5.2.2c–e).
has been reported [125]. Because of the empirical correction terms, the Gaussian and CBS methods are not purely ab initio, except where these terms disappear by subtraction [123]. Some applications of high-accuracy energy methods and suggestions about choosing a method (high accuracy or otherwise) will be given in the following sections (5.5.2.2c–e).
5.5.2.3Thermodynamics; calculating heats of formation
The heat of formation (enthalpy of formation) of a compound is an important thermodynamic quantity, because a table of heats of formation of a limited number of compounds enables one to calculate the heats of reaction (reaction enthalpies) of a great many processes, that is, how exothermic or endothermic these reactions are. The heat of formation of a compound at a specified temperature T is defined [126] as the standard heat ofreaction (standard reaction enthalpy) for formation of the compound at T from its elements in their standard states (their reference states). By the standard state of an element we mean the thermodynamically stablest state at  Pa (standard pressure, about normal atmospheric pressure), at the specified temperature (the exception is phosphorus, for which the standard state is white phosphorus; although red phosphorus is stabler under normal conditions, these allotropes are apparently somewhat ill-defined). The specified temperature is usually 298.15 K (about room temperature). The heat of formation of a compound at room temperature is thus the amount of heat energy (enthalpy) that must be put into the reaction to make the compound from its elements in their normal (room temperature and atmospheric pressure) states; it is the “heat content” or enthalpy of the compound compared to that of the elements. For example,
Pa (standard pressure, about normal atmospheric pressure), at the specified temperature (the exception is phosphorus, for which the standard state is white phosphorus; although red phosphorus is stabler under normal conditions, these allotropes are apparently somewhat ill-defined). The specified temperature is usually 298.15 K (about room temperature). The heat of formation of a compound at room temperature is thus the amount of heat energy (enthalpy) that must be put into the reaction to make the compound from its elements in their normal (room temperature and atmospheric pressure) states; it is the “heat content” or enthalpy of the compound compared to that of the elements. For example,
at 298 K the heat of formation of |
is |
and the heat of formation of |
|
is |
[127]. To make a mole of |
from solid graphite (carbon in |
|
its standard state at 298 K) and hydrogen gas requires –74.87 kJ, i.e. 74.87 kJ are given out – the reaction is mildly exothermic. To make a mole of  from solid graphite and fluorine gas requires –933.20 kJ, i.e. 933.20 kJ are given out – the reaction is strongly
from solid graphite and fluorine gas requires –933.20 kJ, i.e. 933.20 kJ are given out – the reaction is strongly
exothermic. In some sense |
is thermodynamically much stabler with respect to its |
|
elements than is |
Note that the standard heat of formation of an element is zero, |
|
since the reaction in question is the formation of the element from the element, in the same state (no reaction, or a null reaction). Heat offormation is denoted  and heat offormation at, say, 298 K by
and heat offormation at, say, 298 K by  “delta H sub f standard at 298 K”. The delta indicates that this is a difference (enthalpy of the compound minus enthalpy of the elements) and the superscript denotes “standard”.
“delta H sub f standard at 298 K”. The delta indicates that this is a difference (enthalpy of the compound minus enthalpy of the elements) and the superscript denotes “standard”.
There are extensive tabulations of experimentally-determined heats of formation, mostly at 298 K (one way to determine  is from heats of combustion: burning
is from heats of combustion: burning

276 Computational Chemistry
the compound and the elements and measuring calorimetrically the heat evolved enables one to calculate the heat of formation by subtraction).  can also be obtained by ab initio calculations. This is valuable because (1) it is far easier and cheaper than doing a thermochemical experiment, (2) many compounds have not been subjected to experimental determination of their heats of formation, and (3) highly reactive compounds, or valuable compounds available only in very small quantity cannot be subjected to the required experimental protocol. e.g. combustion. Let us see how
can also be obtained by ab initio calculations. This is valuable because (1) it is far easier and cheaper than doing a thermochemical experiment, (2) many compounds have not been subjected to experimental determination of their heats of formation, and (3) highly reactive compounds, or valuable compounds available only in very small quantity cannot be subjected to the required experimental protocol. e.g. combustion. Let us see how  can be calculated.
can be calculated.
Atomization method
Suppose we want to calculate  for methanol. We will calculate the heat offormation at 0 K
for methanol. We will calculate the heat offormation at 0 K and then correct this to 298 K. Figure 5.26 shows the principle behind what has been called the “atomization” method [128]. Methanol is (conceptually) atomized at 0 K into carbon, hydrogen and oxygen atoms(the ground electronic states have been chosen here); it is from this step that the term “atomization” comes. The elements in their normal states are also used to make these atoms, and to make methanol. The heat of formation of methanol at 0 K follows from equating the energy needed to generate the atoms from the elements via methanol
and then correct this to 298 K. Figure 5.26 shows the principle behind what has been called the “atomization” method [128]. Methanol is (conceptually) atomized at 0 K into carbon, hydrogen and oxygen atoms(the ground electronic states have been chosen here); it is from this step that the term “atomization” comes. The elements in their normal states are also used to make these atoms, and to make methanol. The heat of formation of methanol at 0 K follows from equating the energy needed to generate the atoms from the elements via methanol  to that needed to make them directly from the elements in their normal states:
to that needed to make them directly from the elements in their normal states:
i.e.
 is the ab initio atomization energy of methanol, the energy difference between the atoms and methanol. There are a couple points to note about this conceptual scheme. We are converting into carbon atoms graphite, a polymeric material, so strictly speaking Fig. 5.26 should show
is the ab initio atomization energy of methanol, the energy difference between the atoms and methanol. There are a couple points to note about this conceptual scheme. We are converting into carbon atoms graphite, a polymeric material, so strictly speaking Fig. 5.26 should show  where n is a number large
where n is a number large

Ab initio calculations 277
enough to represent the substance graphite rather than just some carbon oligomer. All the species in the figure will then be increased in number by a factor of n, butdivision by this common factor will still give us Eq. (5.188). Another point is that although hydrogen and oxygen are solids at 0 K, we are considering isolated molecules being atomized.
To calculate  we need the 0 K heat of formation of C, H and O atoms, i.e. the atomization energies of graphite, molecular hydrogen, and molecular oxygen, and the 0 K atomization energy of methanol. The atomization energies of hydrogen and oxygen can be calculated ab initio, but not that of graphite, which is a very big “molecule”. For consistency we will use experimental values of all three elemental atomization energies, as recommended [128]. From Eq. (5.176), the 0K atomization energy of methanol is simply the ab initio energies of its constituent atoms minus the ZPE-corrected ab initio of methanol:
we need the 0 K heat of formation of C, H and O atoms, i.e. the atomization energies of graphite, molecular hydrogen, and molecular oxygen, and the 0 K atomization energy of methanol. The atomization energies of hydrogen and oxygen can be calculated ab initio, but not that of graphite, which is a very big “molecule”. For consistency we will use experimental values of all three elemental atomization energies, as recommended [128]. From Eq. (5.176), the 0K atomization energy of methanol is simply the ab initio energies of its constituent atoms minus the ZPE-corrected ab initio of methanol:
Experimental values of  and
and 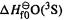 (as well as
(as well as  for other atoms, and references to more extensive tabulations) are given in [113]; in
for other atoms, and references to more extensive tabulations) are given in [113]; in
C 711.2 H 216.035 O 246.6
To calculate  we need (Eq. (5,189))
we need (Eq. (5,189))  for C, H and O atoms (in the states shown) and for methanol. G2 (for comparison with the value in [128]) calculations gave these values (hartrees):
for C, H and O atoms (in the states shown) and for methanol. G2 (for comparison with the value in [128]) calculations gave these values (hartrees):
C |
–37.78430 |
H |
–0.50000 (there are no correlation effects for the H atom; |
|
this is the exact energy) |
O |
–74.98203 |
|
–115.53490 |
From Eq. (5.189)
From Eq. (5.188)
Reference [128] gives the 0 K G2 value by the atomization method as  and the experimental value as (two sources) – 190.7 or
and the experimental value as (two sources) – 190.7 or 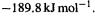
To correct the 0 K heat offormation to that at 298.15 K we add the increase in enthalpy of methanol on going from 0 to 298 K and subtract the corresponding increases for the elements in their standard states. The value for methanol is the difference of two

278 Computational Chemistry
quantities provided in the thermochemical summary at the end of the G2 calculation as implemented in Gaussian 94 or Gaussian 98:
(G2(0 K) is the G2 value for what we have called 
The experimental enthalpy increases for the elements are given in [128];
From these and 
The accepted experimental value [129] is |
|
|
Note that if |
is not wanted, |
can be calculated directly, since from |
Eqs (5.188) and (5.190) the 0 K ab initio energy of the compound is subtracted out and it follows that

Ab initio calculations 279
Formation method
An alternative to the atomization method is what has been called the “formation” method, which is illustrated for methanol in Fig. 5.27. This method utilizes a kind of “pseudo heat of formation”s,  of the compound from atomic carbon and molecular hydrogen and oxygen (the conventional heat of formation is relative to graphite and molecular hydrogen and oxygen). From Fig. 5.27,
of the compound from atomic carbon and molecular hydrogen and oxygen (the conventional heat of formation is relative to graphite and molecular hydrogen and oxygen). From Fig. 5.27,
where the experimental value of  is used, and
is used, and
A calculation using G2 energies gives
The value calculated by this procedure in [128] is  The atomization method usually gives somewhat more accurate heats of formation, at least with the G2-type methods (although for the particular case of methanol this is not so), perhaps
The atomization method usually gives somewhat more accurate heats of formation, at least with the G2-type methods (although for the particular case of methanol this is not so), perhaps
280 Computational Chemistry
because these methods were optimized (via the semiempirical terms, section 5.5.2.2b) to give accurate atomization energies.
Isodesmic reaction method
Finally, heats ofreaction can be calculated by ab initio methods with the aid ofisodesmic reactions (section 5.5.2.2a), as indicated in Fig. 5.28 (actually, the scheme in Fig. 5.28 is not strictly isodesmic – e.g. only on one side of the “isodesmic” equation is there an H–H bond). From this scheme
where
Using G2 values:
With this and the experimental 0 K heats of formation of  and
and  [128]:
[128]:
This is very close to our atomization heat of formation value above  and a little more negative than the experimental value
and a little more negative than the experimental value  [128]).
[128]).
Of the three approaches to calculating heats of formation (atomization, formation and isodesmic), the atomization has been recommended over the formation [128]. The isodesmic (or isodesmic-type, as in Fig. 5.28) should be at least as accurate as the