


336 Computational Chemistry
EASIER QUESTIONS
1.In the term Hartree-Fock, what, essentially, were the contributions of each of these two people?
2.What is a spin orbital? A spatial orbital?
3.At which step in the derivation of the HF energy does the assumption that each electron sees an “average electron cloud” appear?
4.For a closed-shell molecule the number of occupied molecular orbitals is half the number of electrons, but there is no limit to the number of virtual orbitals. Explain.
5.In the simple Hückel method, 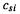 denotes the basis function coefficient for the contribution of atom number s (in whatever numbering scheme we choose) to MO
denotes the basis function coefficient for the contribution of atom number s (in whatever numbering scheme we choose) to MO
number . In the ab initio method, 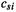 still refers to MO number
still refers to MO number  , but the s does not necessarily denote atom number s. Explain.
, but the s does not necessarily denote atom number s. Explain.
6.The derivation of the Roothaan–Hall equations involves some key concepts: Slater determinant, Schrödinger equation, explicit Hamiltonian operator, energy minimiza-
tion, and LCAO. Using these, summarize the steps leading to the Roothaan–Hall equations 
7.What are the similarities and the differences between the basis set of the extended Hückel method and the ab initio STO-3G basis set?
8.In the simple and extended Hückel methods, the molecular orbitals are calculated and then filled from the bottom up with the available electrons. However, in ab initio calculations the occupancy of the orbitals is taken into account as they are being calculated. Explain. (Hint: look at the expression for the Fock matrix elements in terms of the density matrix.)
9.Isodesmic reactions have been used to investigate aromatic stabilization, but there is not a unique isodesmic reaction for each problem. Write two isodesmic reactions for the ring-opening of benzene, both of which have on each side of the equation the same number of each kind of bond. Have you any reason to prefer one of the equations to the other?
10.List the strengths and weaknesses of ab initio calculations compared to molecular mechanics and extended Hückel calculations. State the molecular features that can be calculated by each method.
HARDER QUESTIONS
1.Does the term ab initio imply that such calculations are “exact”? In what sense might ab initio calculations be said to be semiempirical – or at least not a priori?
2.Can the Schrödinger equation be solved exactly for a species with two protons and one electron? Why or why not?
3.The input for an ab initio calculation (or a semiempirical calculation of the type discussed in chapter 6, or a DFT calculation – chapter 7) on a molecule is usually just the cartesian coordinates of the atoms (plus the charge and multiplicity). So how
Ab initio calculations 337
does the program know where the bonds are, i.e. what the structural formula of the molecule is?
4.Why is it that (in the usual treatment) the calculation of the internuclear repulsion energy term is easy, in contrast to the electronic energy term?
5.In an ab initio calculation on  or
or , one kind of interelectronic interaction does not arise; what is it, and why?
, one kind of interelectronic interaction does not arise; what is it, and why?
6.Why are basis functions not necessarily the same as atomic orbitals?
7.One desirable feature of a basis set is that it should be “balanced.” How might a basis set be unbalanced?
8.In a HF calculation, you can always get a lower energy (a “better” energy, in the sense that it is closer to the true energy) for a molecule by using a bigger basis set, as long as the HF limit has not been reached. Yet a bigger basis set does not necessarily give better geometries and better relative (i.e. activation and reaction) energies. Why is this so?
9.Why is size-consistency in an ab initio method considered more important than variational behavior (MP2 is size-consistent but not variational)?
10.A common alternative to writing a HF wavefunction as an explicit Slater determinant is to express it using a permutation operator 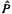 which permutes (switches) electrons
which permutes (switches) electrons
around in MOs. Examine the Slater determinant for a two-electron closed-shell molecule, then try to rewrite the wavefunction using 
This page intentionally left blank
Chapter 6
Semiempirical Calculations
Current “ab initio” methods were limited to very inaccurate
calculations for very small molecules.
M J. S. Dewar, A Semiempirical Life, 1992.
6.1 PERSPECTIVE
We have already seen examples of semiempirical (SE) methods, in chapter 4: the simple Hückel method (SHM, Erich Hückel, ca. 1931) and the extended Hückel method (EHM, Roald Hoffman, 1963). These are semiempirical (semiexperimental) because they combine physical theory with experiment. Both methods start with the Schrödinger equation (theory) and derive from this a set of secular equations which may be solved for energy levels and molecular orbital coefficients (most efficiently by diagonalizing a Fock matrix; see chapter 4). However, the SHM gives energy levels in units of a parameter  that can be translated into actual quantities only by comparing SHM results with experiment, and the EHM uses experimental ionization energies to translate the Fock matrix elements into actual energy quantities. SE calculations stand in contrast to empirical methods, like molecular mechanics (MM, chapter 3), and theoretical methods, like ab initio calculations (chapter 5). MM starts with a model of a molecule as balls and springs, a model that works but whose theoretical justification lies outside MM. The ab initio method, like the Hückel methods, starts with the Schrödinger equation but does not appeal to experiment (beyond invoking, when actual quantities are needed, experimental values for Planck’s constant, the charge on the electron and proton, and the masses of the electron and atomic nuclei – fundamental physical constants which could be calculated only by some deep theory of the origin and nature of our universe [1].
that can be translated into actual quantities only by comparing SHM results with experiment, and the EHM uses experimental ionization energies to translate the Fock matrix elements into actual energy quantities. SE calculations stand in contrast to empirical methods, like molecular mechanics (MM, chapter 3), and theoretical methods, like ab initio calculations (chapter 5). MM starts with a model of a molecule as balls and springs, a model that works but whose theoretical justification lies outside MM. The ab initio method, like the Hückel methods, starts with the Schrödinger equation but does not appeal to experiment (beyond invoking, when actual quantities are needed, experimental values for Planck’s constant, the charge on the electron and proton, and the masses of the electron and atomic nuclei – fundamental physical constants which could be calculated only by some deep theory of the origin and nature of our universe [1].
The Hückel methods were discussed in chapter 4 rather than here because extensive application of these methods came before widespread use of ab initio methods, and because the simple Hückel, extended Hückel and ab initio methods form a conceptual progression in which the first two methods aid understanding of the next one in this