

Нарушения межуточного обмена углеводов
Одним из этапов обмена углеводов в организме является межуточный обмен – окисление углеводов в тканях организма до конечных продуктов – СО2 и Н2О. Процесс окисления Гл идет по двум основным путям: анаэробный гликолиз и аэробный гликолиз (рис. 2).
Распад Гл в анаэробных условиях и при непрямом превращении протекает почти одинаково до образования пировиноградной кислоты (ПК).
В ходе 11 последовательных реакций, необходимых для того, чтобы завершилось это превращение, образуется ряд промежуточных продуктов, представляющих собой эфиры фосфорной кислоты (фосфаты). Их фосфатная группа переносится на аденозиндифосфат (АДФ) с образованием АТФ. Чистый выход АТФ составляет 2 молекулы АТФ на каждую молекулу глюкозы.
В анаэробных условиях ПК восстанавливается в молочную кислоту (МК), которая в печени участвует в глюконеогенезе и ресинтезируется через цикл Кори в Гл. В крови здоровых людей содержание МК составляет 0,6-1,7 ммоль/л.
В ходе эволюции развился механизм, обеспечивающий полное окисление глюкозы до CO2 и воды – аэробный процесс, в котором чистый выход АТФ составляет 38 молекул АТФ на каждую окисленную молекулу глюкозы (рис. 3). Этот процесс потребления клетками кислорода для образования богатых энергией соединений известен как клеточное дыхание (аэробное). В отличие от анаэробного процесса, осуществляемого ферментами цитоплазмы, окислительные процессы протекают в митохондриях.
В митохондриях пировиноградная кислота окисляется до СО2 в шести последовательных реакциях, в каждой из которых пара электронов переносится на общий акцептор – кофермент никотинамидадениндинуклеотид (НАД). Эту последовательность реакций называют циклом трикарбоновых кислот, циклом лимонной кислоты или циклом Кребса. Из каждой молекулы глюкозы образуется 2 молекулы пировиноградной кислоты; 12 пар электронов отщепляется от молекулы глюкозы в ходе ее окисления.
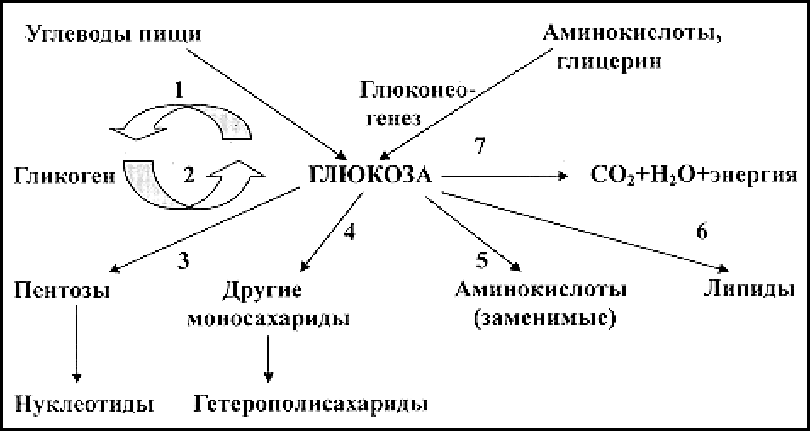
Рис. 2. Основные этапы межуточного обмена углеводов.
– гликогенез
– гликогенолиз
– пентозо-фосфатный путь (образование пентоз)
– образование гетерополисахаридов
– образование заменимых аминокислот
– образование триглицеридов
– аэробный (до CO2 и Н2О) и анаэробный (до пирувата (ПК) и лактата (МК)) пути
В аэробных условиях при участии пируватдегидрогеназного комплекса и 5 коферментов (тиаминдифосфата, рибофлавина, пантотеновой и липоевой кислот, никотинамида) происходит окисление пировиноградной кислоты до ацетил-КoА, который затем подвергается дальнейшим превращениям в цикле Кребса, конечными продуктами которого является СО2, Н2О.
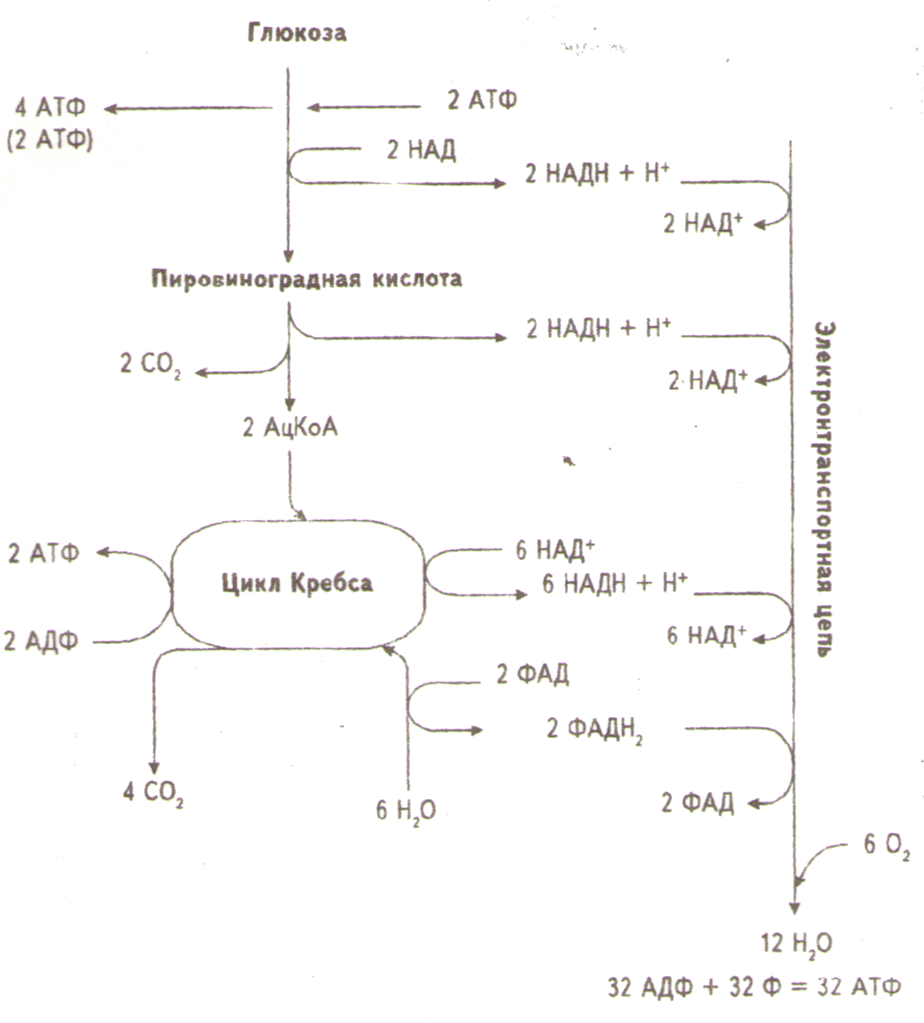
Рис.3. Образование АТФ при полном окислении глюкозы
Печень обладает свойством синтезировать глюкозу из других сахаров, например фруктозы и галактозы, или из других продуктов промежуточного метаболизма. Превращение лактата в глюкозу в цикле Кори и аланина в глюкозу в глюкозо-аланиновом цикле играет особую роль в обеспечении глюкозой эритроцитов и мышечных клеток.
Синтез глюкозы de novo (до 250 г в сутки) происходит в основном в печени. Процесс глюконеогенеза может идти в почках, однако из-за небольших размеров почек их вклад в синтез глюкозы составляет всего 10%.
Глюконеогенез контролируется гормонами. Кортизол, глюкагон и адреналин стимулируют этот процесс, а инсулин, напротив, подавляет.
В печени наиболее важными субстратами в глюконеогенезе являются лактат, поступающий из мышечной ткани и эритроцитов, аминокислоты – из желудочно-кишечного тракта (глюкогенные аминокислоты) и мышц (аланин), а также глицерин – из жировой ткани. В почках основным субстратом являются аминокислоты (глутамин и другие), а также лактат, глицерин и фруктоза, поступающие из крови. Так как почки одновременно потребляют глюкозу, общий баланс глюкозы сохраняется без изменений.
Независимо от процесса глюконеогенеза в почках идет процесс реабсорбции глюкозы из первичной мочи. Это энергозависимый процесс, сопряженный с гидролизом АТФ. Вместе с тем он сопровождается сопутствующим транспортом ионов Na+ (по градиенту, так как концентрация Na+ в первичной моче выше, чем в клетках). Этот процесс получил название «вторичного активного транспорта».
Табл. 1. Основные клинико-биохимические показатели крови,
характеризующие обмен углеводов
|
Показатель |
нормы в системе СИ |
|
глюкоза плазмы натощак |
3,3-6,4 mМ (3,3 – 5,5 mM в крови)
|
|
пируват (пировиноградная кислота) |
56,8-113,6 mМ
|
|
лактат |
0,6-1,7 mМ
|
Табл.2. Показатели углеводного обмена в сыворотке крови у детей различного возраста
|
Показатель |
возраст | ||
|
новорожденные |
1 мес – 1 год |
2–14 лет | |
|
глюкоза мм/л |
2,87 4,44-6,67 |
3,0-5,28 |
3,5-5,0 |
|
пируват, мкм/л |
171-319 |
57-125 |
46-125 |
|
лактат мМ/л |
2,0-2,44 |
1,33-1,78 |
1,0-1,67 |
Нарушения межуточного обмена углеводов касаются, прежде всего, аэробного пути обмена, в результате чего в организме накапливаются пируват и лактат, нарушается цикл Кребса.
Причинами нарушения межуточного обмена углеводов являются: нарушение функции поджелудочной железы, недостаток коферментов (витамина B1), дефицит кислорода (различные виды гипоксий).
Нарушение этапа синтеза и распада гликогена в организме
Углеводы, поступая в большом количестве, в желудочно-кишечном тракте гидролизуются до глюкозы или иных сахаров, которые затем в печени превращаются в глюкозу. Здесь из глюкозы синтезируется гигантский полимер гликоген путем присоединения друг к другу остатков глюкозы с отщеплением молекул воды (число остатков глюкозы в молекулах гликогена доходит до 30 000) (рис.4).
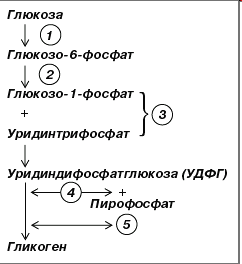
Рис.4. Синтез гликогена
1– гексокиназа или глюкокиназа;
2 – фосфоглюкомутаза;
3 – глюкозо-1-фосфат-уридилилтрансфераза (УДФГ-пирофосфорилаза);
4– гликогенсинтаза;
5– гликогенветвящийся фермент или амило-(1->4) -> (1->6)-трансглюкозидаза.
Гликоген животных, как и амилопектин растений, представляет собой разветвленный гомополимер глюкозы, в котором остатки глюкозы соединены α(1→4)-гликозидной связью (рис.5). Связи в точках ветвления находятся в положении α(1→6) примерно каждого 10-го остатка. Таким образом, возникает древовидная структура с молекулярной массой >1ּ107 Да (до 50 000 остатков).
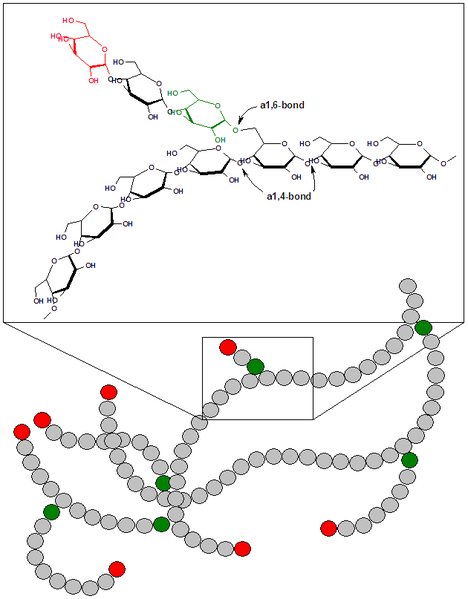
Рис.5 Химическая структура гликогена.
Гликоген служит резервом углеводов, из которого по мере метаболической потребности могут высвобождаться глюкозофосфат или глюкоза.
Гликоген печени никогда не расщепляется полностью. Как правило, укорачиваются или удлиняются (при высоком содержании глюкозы) только невосстанавливающиеся концы древовидной структуры. Удлинение цепи катализируется гликоген-синтазой.
Разветвленная структура гликогена облегчает быстрое освобождение углеводных остатков. Наиболее важным ферментом деградации гликогена является гликоген-фосфорилаза, отщепляющая от невосстанавливающего конца цепи остатки глюкозы в виде глюкозо-1-фосфата.
В организме человека может содержаться до 450 г гликогена, треть из которого накапливается в печени, а остальное – главным образом в мышцах. Содержание гликогена в других органах незначительно. Гликоген представляет форму легко мобилизуемого депо гликогена, восполняющего дефицит глюкозы в крови. Гликоген печени служит прежде всего для поддержания уровня глюкозы в крови в период между приемом пищи и всасыванием глюкозы в кровь. Поэтому содержание гликогена в печени варьирует в широких пределах. При длительном голодании оно падает почти до нуля, после чего начинается снабжение организма глюкозой с помощью глюконеогенеза. Гликоген мышц служит резервом энергии и не участвует в регуляции уровня глюкозы в крови. В мышцах отсутствует глюкозо-6-фосфатаза, поэтому гликоген мышц не может быть источником глюкозы в крови, в связи с чем колебания содержания гликогена в мышцах меньше, чем в печени.
Когда возникает потребность в энергии, гликоген вновь распадается до глюкозы в реакции, продуктом которой является глюкозофосфат (рис.6).
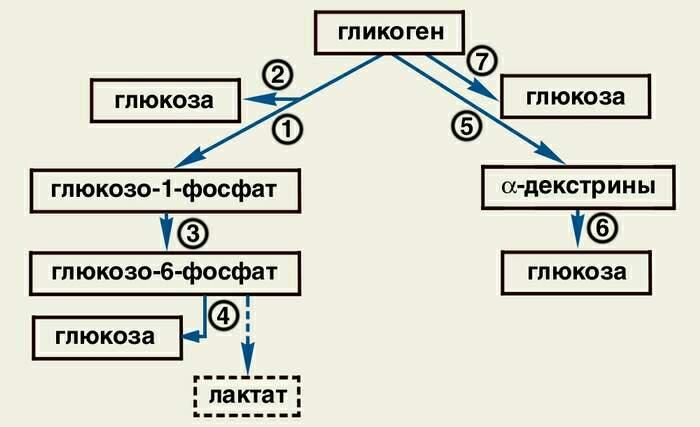
Рис. 6. Гликогенолиз.
1 – фосфорилаза;
2 – амило-1,6-глюкозидаза;
3 – фосфоглюкомутаза;
4 – глюкозо-6-фосфатаза;
5 – α-амилаза;
6 – нейтральные α-глюкозидазы;
7 – кислая α-глюкозидаза
Этот глюкозофосфат направляется на путь гликолиза – процесса, составляющего часть пути окисления глюкозы. В печени глюкозофосфат может также подвергнуться гидролизу и образующаяся глюкоза поступает в кровоток и доставляется кровью к клеткам в разных частях тела.
Гликогеновые болезни – (glycogenosis, единственное число; гликоген + -sis; синоним: болезнь накопления гликогена, гликогеновая болезнь) – группа наследственных болезней, которые обусловлены недостаточностью ферментов, участвующих в обмене гликогена; характеризуются нарушением структуры гликогена, недостаточным или избыточным накоплением его в различных органах и тканях. Распространенность гликогенозов в популяции составляет 1:68000 – 1:40000.
Агликогенозы – невозможность образования гликогена вследствие дефицита фермента гликогенсинтетазы. При агликогенозе, характеризующемся резким снижением запасов гликогена в печени, наблюдаются гипогликемические состояния вплоть до развития комы. Кома может возникать вскоре после рождения при позднем прикладывании ребенка к груди матери, в более старшем возрасте – в перерывах между кормлениями, утром натощак. Если ребенок выживает, в последующем при отсутствии лечения нарушается психомоторное развитие. Гипогликемия не купируется введением глюкагона, после применения глюкозы длительно сохраняется гипергликемия. На фоне гипогликемии отмечается гиперкетонемия, уровень лактата в крови нормальный. Тип наследования не установлен.
Гликогенозы – заболевания, обусловленные дефектом ферментов, участвующих в распаде гликогена.
По характеру ферментативной недостаточности выделяют 11 типов гликогенозов (табл.3). В зависимости от преобладания симптомов поражения печени или мышц условно выделяют печеночные и мышечные формы гликогенозов.
Табл.3. Гликогенозы
|
Тип гликогеноза |
Фермент с нарушенной активностью |
Основные органы, ткани и клетки, в которых найден дефект фермента |
|
1-й тип (болезнь Гирке) |
глюкозо-6-фосфатаза |
печень, почки, слизистая оболочка тонкой кишки |
|
2-й тип (болезнь Помпе) |
альфа-1,4-глюкозидаза |
печень, почки, селезенка, мышцы, нервная ткань, лейкоциты |
|
3-й тип (болезнь Кори) |
амило-1,6-глюкозидаза |
печень, мышцы, лейкоциты, эритроциты |
|
4-й тип (болезнь Андерсена) |
D-1,4-глюкано-α-глюкозилтрансфераза |
печень, мышцы, почки, лейкоциты |
|
5-й тип (болезнь МакАрдля) |
гликогенфосфорилаза |
мышцы |
|
6-й тип (болезнь Гирса) |
гликогенфосфорилаза гепатоцитов |
печень, лейкоциты |
|
7-й тип (болезнь Томпсона) |
фосфоглюкомутаза |
мышцы, эритроциты |
|
8-й тип (болезнь Таруи) |
фосфофруктокиназа |
мышцы, эритроциты, головной мозг, печень |
|
9-й тип (болезнь Хага) |
киназы фосфорилаза в гепатоцитах |
печень |
|
10-й тип |
ц-АМФ-зависимая киназа фосфорилазы |
печень, мышцы |
|
11-й тип |
фосфоглюкомутаза? |
печень, почки |
Гликогенозы 0, I, III, IV, VI, VIII, IX, X и XI типов относят к печеночным, V и VII типов – к мышечным. Гликогеноз II типа проявляется поражением многих органов и систем (генерализованная форма) или только мышц. Возможно сочетание гликогенозов нескольких типов (например, I и III типа).
Гликогеноз I типа (болезнь Гирке, гепатонефромегальный Г.) наследуется по аутосомно-рецессивному типу. Первые проявления – отсутствие аппетита, рвота, гипогликемические судороги (комы), респираторный дистресс-синдром, интермиттирующее повышение температуры тела, гепатомегалия, нефромегалия, стеаторея, кетонурия – выявляются сразу после рождения или в грудном возрасте. С течением времени прогрессируют гепатомегалия и нефромегалия за счет гликогенной инфильтрации. Характерны отставание в росте, диспропорция тела (большая голова, короткие шея и ноги), кукольное лицо, гипотония мышц. В связи с резкой гипогликемией больные вынуждены почти постоянно принимать пищу.
Часто присоединяющиеся вторичные инфекции приводят к значительному усилению кетоацидоза и гипогликемии и нередко являются причиной смерти. Иногда развиваются геморрагический синдром, кожный ксантоматоз. Нервно-психическое развитие удовлетворительное. Половое созревание значительно задерживается. Состояние больных несколько улучшается в пубертатном периоде. Биохимические нарушения: гипогликемия, кетоз, гиперлактацидемия, гиперлипемия, повышение в крови уровня неэтерифицированных жирных кислот, гликогена, холестерола, мочевой кислоты, нарушение почечного клиренса для ряда веществ. Введение адреналина, глюкагона, галактозы вызывает значительную гиперлактацидемию, но не гипергликемию, т.к. глюкозо-6-фосфатаза в печени отсутствует.
Генерализованная форма гликогеноза II типа (болезнь Помпе) наследуется по аутосомно-рецессивному типу. Первые симптомы болезни выявляются через несколько дней или недель (до 6 мес.) после рождения. Отмечаются цианоз (общий и интермиттирующий), расстройство дыхания (ускоренное, поверхностное), беспокойство или адинамия. Постепенно увеличивается язык (макроглоссия), нарастает мышечная гипотония. Отмечаются отсутствие аппетита, пилороспазм, задержка роста. Увеличиваются размеры печени, селезенки, почек, сердца. В связи с гипертрофией миокарда сердце приобретает шаровидную форму, появляются изменения ЭКГ. Часто возникают бронхиты, ателектазы легких, гипостатические пневмонии. Наблюдаются миодистрофия, гипорефлексия, бульбарные нарушения, спастические параличи. В сыворотке крови повышены содержание мочевой кислоты, активность трансаминаз и альдолазы.
Мышечная форма гликогеноза II типа возникает при дефиците кислой -1,4-глюкозидазы только в мышцах. В этих случаях болезнь, как правило, проявляется в более позднем возрасте и по клинической картине напоминает миопатии.
Гликогеноз III типа (болезнь Кори, болезнь Форбса) наследуется по аутосомно-рецессивному типу. Наблюдаются гепатомегалия с первых месяцев жизни, мышечная гипотония, гипертрофия отдельных мышечных групп. У некоторых больных отмечаются гипертрофия миокарда, нарушения сердечной проводимости и кровообращения. При биохимических исследованиях выявляют гипогликемию натощак, кетоз, липемию, повышение уровня гликогена в эритроцитах. После 5-летнего возраста и особенно в пубертатном периоде развитие заболевания значительно замедляется.
Гликогеноз IV типа (болезнь Андерсена, амилопектиноз, диффузный гликогеноз с циррозом печени) проявляется с первых месяцев жизни и характеризуется гепатосплено-мегалией, развитием цирроза печени, желтухой, гипогликемией. Больные обычно погибают на первом году жизни). Болезнь передается предположительно по аутосомно-рецессивному или связанному с полом типу.
Гликогеноз V типа (болезнь Мак-Ардла, миофосфорилазная недостаточность) наследуется по аутосомно-рецессивному типу. Лица мужского пола болеют в 5 раз чаще. В связи с гликогенной инфильтрацией скелетные мышцы увеличиваются в объеме, становятся очень плотными. Мышечная слабость, мышечные спазмы, тахикардия при физической нагрузке появляются в первые десять лет жизни и прогрессируют. Наблюдается транзиторная миоглобинурия. Концентрация лактата в крови после физической нагрузки уменьшается.
Гликогеноз VI типа (болезнь Герса, гепатофосфорилазная недостаточность) наследуется предположительно по аутосомно-рецессивному типу. Проявляется обычно на первом году жизни. Характерны значительное увеличение печени в результате гликогенной инфильтрации гепатоцитов, задержка роста, кукольное лицо, гиперлипемия, гипергликемия после внутривенного введения галактозы, повышенное содержание гликогена в эритроцитах.
Гликогеноз VII типа (болезнь Таруи, миофосфофруктокиназная недостаточность) по клиническим проявлениям сходен с гликогенозом V типа. Характерны мышечная слабость, утомляемость и отсутствие гиперлактацидемии после физической нагрузки.
Гликогеноз VIII типа (болезнь Томсона) встречается крайне редко. После рождения постепенно увеличиваются размеры печени, затем появляются нистагм («танцующие глаза»), атаксия. Неврологическая симптоматика прогрессирует вплоть до развития мышечной гипертонии, децеребрации. Больные, как правило, погибают. Тип наследования не установлен.
Гликогеноз IX типа (болезнь Хага) наследуется по рецессивному, связанному с полом типу. У больных наблюдается гепатомегалия. Другие симптомы, характерные для печеночных форм Г, не выражены.
Гликогеноз Х типа описан у единственного больного. Наблюдалась гепатомегалия, через 6 лет после начала заболевания появились мышечные боли и спазмы мышц после физических упражнений. Тип наследования не установлен.
Гликогеноз XI типа характеризуется значительным увеличением печени и резкой задержкой роста. Активность трансаминаз и уровень липидов в сыворотке крови могут быть повышены, содержание фосфатов снижено. Характерны генерализованная гипераминоацидурия, галактозурия, глюкозурия, фосфатурия. Наблюдаются симптомы гипофосфатемического рахита. В пубертатном периоде возможны уменьшение размеров печени, ускорение роста, нормализация уровня фосфора в крови. Тип наследования не установлен.
Для подтверждения диагноза гликогеноза и установления его типа в стационаре проводят биопсию печени, мышц (иногда кожи) с последующим гистохимическим исследованием; при этом определяют содержание гликогена в тканях и активность ферментов, участвующих в его обмене. При гликогенозе II типа определяют также активность кислой α-1,4-глюкозидазы в форменных элементах крови, а также в культуре клеток фибробластов кожи и мышц больного. Гликогеноз II типа можно диагностировать пренатально путем биохимического исследования клеток слущивающегося эпителия кожи плода, находящихся в околоплодной жидкости, полученной с помощью амниоцентеза.
Лечение направлено на борьбу с обменными нарушениями, в т.ч. с ацидозом. В некоторых случаях эффективно применение глюкагона, анаболических гормонов и глюкокортикоидов. Частые приемы пищи с высоким содержанием легкоусвояемых углеводов необходимы при гипогликемии. При мышечных формах Г. улучшение отмечается при соблюдении диеты с высоким содержанием белка, назначении фруктозы (внутрь по 50–100 г в день), поливитаминов, АТФ. Имеются сообщения о хирургическом лечении I и III типов Г. (портокавальная транспозиция сосудов, перевязка портальной вены и наложение анастомоза конец в бок между воротной и нижней полой венами). Предпринимаются попытки введения больным недостающих ферментов.
Прогноз для жизни неблагоприятный при гликогенозах 0, I, II (генерализованной форме), IV и VIII типов; смерть нередко наступает на первом году жизни, особенно при развитии интеркуррентных заболеваний. В остальных случаях прогноз для жизни, как правило, благоприятный, выздоровление невозможно.
Кроме Г существуют другие формы нарушения углеводногообмена, обусловленные генетическими дефектами синтеза отдельных ферментов важных путей метаболизма углеводов (галактоземия, фруктозурия, непереносимость лактозы и другие заболевания).