

«Опис фенотипу»







ЗМІСТОВИЙ МОДУЛЬ 4.
Моногенні хвороби
Тема: Загальна характеристика моногенної патології.
Клініка і генетика окремих форм
моногенних хвороб.
Основні питання:
1.
Моногенні хвороби.
![]()
Визначення поняття. Етіологія та класифікація.
2. Загальні закономірності патогенезу моногенної патології.
3. Головні риси клінічної картини моногенної патології.
4. Клінічний поліморфізм моногенної патології та його причини.
5. Генетична гетерогенність моногенних захворювань.
6. Клініка, генетика та діагностика нейрофіброматозу.
7. Клініка, генетика та діагностика вродженого гіпотиреозу.
Клініка, генетика та діагностика фенілкетонурії.
Клініка, генетика та діагностика муковісцидозу.
10.Клініка, генетика та діагностика синдрому Марфана.
Клініка, генетика та діагностика гомоцистинурії.
Клініка, генетика та діагностика адреногенітального синдрому.
Клініка, генетика та діагностика синдрому Елерса-Данлоса.
Клініка, генетика та діагностика онкогенетичних синдромів.
Геномний імпринтинг. Визначення поняття.
Хвороби геномного імпринтингу. Етіологія, патогенез, клінічні форми.
Моногенні хвороби або генні хвороби (така назва розповсюджена закордоном) – це різноманітна за клінічними проявами група захворювань, які обумовлені мутаціями на генному рівні та підлягаються менделевському успадкуванню. В основі цієї групи спадкової патології знаходяться одиничні генні або крапкові мутації, які включають дефекти екзотів (делеції, вставки, заміни, інверсії), дефекти інтронів та фланкіруючих ділянок (заміна у сигналі поліаденілірівання), що призводить до зміни складу та порядку нуклеотидів у молекулі ДНК, порушенню трансляції генетичної інформації від ДНК до РНК, від РНК на рибосоми та до змін послідовності амінокислот у поліпептиді.
На теперішній час відомо більш 5000 нозологічних одиниць моногенних хвороб. У різних країнах їх виявляють у 30-65 дітей у розрахунку на 1000 новонароджених, що складає 3,0 – 6,5 %, а у структурі загальної смертності дітей до 5 років на їх долю приходиться 10-14%.
Моногенна патологія займає значне місце у сучасній медицині. У залежності від того, яка система найбільш вражена, умовно виділяють спадкові хвороби шкіри, очей, нервової системи, ендокринні, опорно-рухового апарату, нервово-м’язової системи, крові, сердцево-судинної системи, шлунково-кишкового тракту, моче полової системи та інші. Для деяких груп хвороб існують навіть спеціальні терміни: нейрогенетика, онкогенетика, офтальмогенетика, дерматогенетика тощо. Умовність такої класифікації не визиває сумнівів, тому що у деяких хворих ті самі хвороби проявляються по-різному. Наприклад, муковісцидоз може протікати з переважним враженням шлунково-кишкового тракту або легенів.
Патогенетична класифікація генних хвороб підрозділяє їх в залежності від того, на що спрямований основний патогенетичний ланцюг, який може призвести к порушенню обміну речовин, аномаліям морфогенезу або комбінації першого та другого. При цоьму виділяють:
спадкові хвороби обміну (СХО),
вроджені вади розвитку (моногенної природи),
комбінірованні стани.
СХО в свою чергу підрозділяють за типом порушеного обміну:
вуглеводів,
амінокислот,
вітамінів,
липидів,
металів та інш.
Тип успадкування моногенного захворювання пов’язаний з локалізацією патологічного гена в аутосомі або статевій хромосомі та залежить від його домінантності або рецесивності. При цьому чоловіки та жінки вражаються с однаковою частотою при аутосомному типі успадкування та з різною частотою при зчепленому зі статтю успадкуванні. Виділяють:
аутосомно-домінантні хвороби: ахондроплазія, недосконалий остеогенез, нейрофіброматоз, ретінобластома, сімейна гіперхолестерінемія, синдром Марфана, хорея Гентингтона та ін.;
аутосомно-рецесивні хвороби: муковісцидоз, фенілкетонурія, адрено-генітальний синдром, альбінізм, атаксія-телеангектазія, галактоземія;
Х-зчеплениі домінантні хвороби: гіпофосфатемія, синдром Гольтца-Горліна;
Х-зчеплениі рецесивні хвороби: міодистрофія Дюшенна-Беккера, гемофілія, синдром Мартина – Белл.
Прикладами поширених моногенних хвороб є наступні нозологічні одиниці:
Муковісцидоз (cystic fibrosis – кистофиброз). Ген локализується у сегменті 7q32 и кодує білок – регулятор трансмебранної проводимості (CFTR). Частота для країни Європы та Північної Америки 1 : 2000.
Фенілкетонурія (ФКУ). При ФКУ, що обумовлена дефіцитом дігідроптеридинредуктази, ген локализується у 14q15.1. Частота – 1 : 10 000; у деяких популяціях 1 : 1000.
Міодистрофія Дюшена-Беккера (Xp21) – 1:3000–3500 для чоловіків.
Нейрофіброматоз Реклингаузена, тип 1 (17q11.2) и аказуістичний тип 2 (22q11.2) – 1 : 3000–5000.
Врождений гіпотеріоз (8q24.3) – 1 : 4700. При незобових формах гіпотеріозу гени локализуються у сегментах 1р13 и 14q31.
Синдром Мартина–Белл (фрагільна X-хромосома або зчеплена з Х-хромосомою розумова відстілість). Ген захворювання (FMR1) локализується у сегменті Xq27 (хромосомний маркер – fra Xq27.3). Частота у популяції від 0,3 до 1,0 на 1000.
Моногенні хвороби за фенотипичними проявами поділяються на ферментопії (хвороби обміну речовин, у т.ч. хвороби, що обумовлені порушенням репарації ДНК): хвороби, що обумовлені молекулярною патологією структурних білків; имунопатологією, у т.ч. хворобами, викликаними порушеннями в системі комплементу; порушеннями синтезу транспортних білків (у т.ч. білків крові) і пептидних гормонів; патологією згортувальної системи крові; дефектами механізму переносу речовин через клітинні мембрани.
Серед моногенних хвороб також виділяють групу синдромів із МПВР. Більшість відомих спадкових хвороб обумовлені мутаціями структурних генів, можливість етиологичной ролі мутацій генів-регуляторів при деяких захворюваннях дотепер доведена лише частково.
Ферментопатії, що відносяться до моногенних хвороб по фенотипичним проявам, становлять найбільш велику й найкраще вивчену групу спадкових захворювань. Первинний дефект ферменту розшифрований більш ніж для 200 ферментопатій. Можливі наступні причини ферментопатій:
фермент не синтезується зовсім;
у молекулі ферменту порушена послідовність амінокислот, тобто змінена його первинна структура;
відсутній або неправильно синтезується кофермент відповідного ферменту;
активність ферменту змінена у зв'язку з аномаліями в інших ферментних системах;
блокада ферменту обумовлена генетично детермінованим синтезом речовин, що викликають інактивацію цього ферменту.
Мутація гену може викликати порушення синтезу білків, що виконують пластичні (структурні) функції. Порушення синтезу структурних білків - імовірна причина таких захворювань, як остеодисплазії й остеогенез недосконалий. Є дані про певну роль цих порушень у патогенезі спадкових нефритоподібних захворюваннях - синдромі Альпорта й сімейної гематурії. Дисплазія тканин в результаті аномалій у структурі білків може спостерігатися не тільки в нирках, але й у будь-яких інших органах. Патологія структурних білків характерна для більшості спадкових хвороб, що успадковуються по аутосомно-домінантному типі.
Мутація гену може привести до розвитку хвороб, викликаних иммунодефіцитними станами. Найбільше важко протікає агаммаглобулинемія, особливо в сполученні з аплазією тимусу.
Причиною появи гемоглобіну з аномальною структурою при серповидно-клітинній анемії є заміна в його молекулі залишку глутаминової кислоти на залишок валину. Подібна заміна є наслідком генної мутації. Це відкриття послужило початком інтенсивного вивчення великої групи моногенних хвороб, названих гемоглобинопатіями.
Відомий ряд мутантних генів, що контролюють синтез факторів згортання крові. Генетично детерміновані порушення синтезу антигемофиличнго глобуліну (фактор VІІІ) приводять до розвитку гемофілії А. При порушенні синтезу тромбопластичного компоненту (фактор ІX) розвивається гемофілія В. Недолік попередника тромбопластину лежить в основі патогенезу гемофілії С.
Генні мутації можуть бути причиною порушення механізму транспорту різних з'єднань через клітинні мембрани. Найбільш вивчені спадкова патологія транспорту амінокислот у кишечнику і нирках, синдром мальабсорбції глюкози й галактози. Прикладом захворювання, викликаного генетично обумовленим дефектом механізму транспорту амінокислот через клітинні мембрани, є цистинурія, клінічним проявом якої є нефролітіаз і симптоми пієлонефриту. Класична цистинурія обумовлена порушенням переносу ряду діаминокарбонових кислот (аргініну, лізину) і цистину через клітинні мембрани як у кишечнику, так і в нирках. Патологія реабсорбції глюкози в ниркових канальцах, ниркова глюкозурія, пов'язана з порушенням функції мембранних білків-переносників або з дефектами в системі забезпечення енергією процесів активного транспорту глюкози, успадковується по аутосомно-домінантному типі. Порушення реабсорбції бікарбонатів у проксимальних відділах ниркових канальців або порушення секреції Н-іонів клітками ниркового епітелію дистальних відділів ниркових канальців лежить в основі розвитку двух типів нирково-канальцевого ацидозу.
Експресія генів, що записані в генетичному коді, регулюється процесом метилювання, а останній впливає на генетичний код через збільшення генних мутацій, внаслідок дезамінування цитозину. Мутації генів, які контролюють епігенетичну модифікацію, приводять до епігенетичних хвороб, які поділяють на дві групи. До першої групи віднесені хвороби із порушенням епігенетичного статусу окремих ділянок геному (локальний епігенетичний ефект), до другої – хвороби із порушенням епігенетичного статусу всього геному (глобальний епігенетичний ефект).
До першої групи увійшли хвороби геномного імпринтингу і хвороби, пов'язані із порушеним статусом метилювання окремих генів внаслідок мутацій "de novo", які виникають в соматичних клітинах. До другої групи відносять гетерохроматинові хвороби, які обумовлені мутаціями або варіантами генів, продукти яких втягнуті у підтримку рівня метилювання ДНК та модифікацію структури гетерохроматину (наприклад, синдром Ретта, Рубінштейна-Тейбі, Коффіна-Лоурі).
Однім з найвідоміших епігенетичних процесів є геномний імпринтинг (від англ. imprint — відбиток), який відповідає за диференційне маркування материнських та батьківських гомологічних хромосом, що призводить до різних фенотипових проявів успадкованих від матері або батька мутацій у нащадків. У ділянках геному, що підвладні геномному імпринтингу, експресується лише один із двох алелей – батьківській або материнський, а другий алель в наслідок присутності на ньому деякого „відбитка” є імпринтированим (вилученним). Цей спосіб регуляції роботи генів є свідченням нееквивалентного вкладу батьків у геном нащадків.
У теперішній час відомо біля 30 імпинтированих генів у людини, що призводять до розвитку унібатьківської дисомії та відповідають за розвиток аномальних фенотипів при синдромах Прадера-Виллі, Ангельмана, Відемана-Беквіта, Рассела-Сильвера, а також деяких злоякісних новоутворювань (рабдоміосаркома, остеосаркома, пухлина Вільмса). Окрім цього, внаслідок ізодисомії виникає гомогенізація генів, що сприяє проявам рецесивних моногенних дефектів. Таким чином проблема діагностики проявів геномного імпринтингу є надзвичайно важливою у питанні розуміння суті цих спадково обумовлених епігенетичних захворювань.
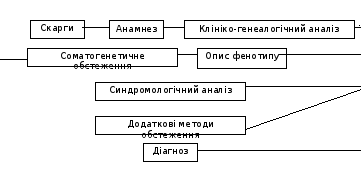
Граф логічної структури теми