

Методы определения порядка реакций
Так как стадии, через которые протекают реакции, в большинстве случаев неизвестны, предвидеть порядки реакции по каждому веществу невозможно; их следует определять специальными методами, которые будут рассмотрены ниже.
Для определения порядка реакции в целом необходимо сначала установить порядок реакции по каждому веществу, вступающему в реакцию. Сумма порядков реакции по каждому веществу дает порядок реакции в целом. Чтобы определить порядок реакции по данному веществу, необходимо создать такие условия, при которых в процессе реакции изменялась бы концентрация только этого вещества. Для этого концентрации всех остальных участников реакции должны быть настолько большими, чтобы изменением их во времени можно было пренебречь и значения этих концентраций можно было бы ввести в константу скорости (метод изолирования Оствальда). Тогда для реакции
1 A + 2 B + 3 C = 4 D + 5 E ,
протекающей при V = const, можно записать
–
![]() =
k1
=
k1
![]() , где k1
= k
, где k1
= k
![]()
![]() .
(1)
.
(1)
Все известные методы определения частных порядков реакций можно разделить на две группы: интегральные и дифференциальные.
Интегральные методы. Здесь используются кинетические уравнения для скорости реакции в интегральной форме (полученные после интегрирования дифференциального уравнения скорости реакции).
1. По времени полураспада (см. «Необратимые реакции»).
Для реакции 1-го порядка не зависит от начальной концентрации: = 0,693/ k ;
для реакции 2-го порядка обратно пропорционально начальной концентрации исходного вещества: = 1/ ka ;
для реакции 3-го порядка: = 3/(2ka 2).
В общем случае время полураспада обратно пропорционально an–1. Проводят несколько опытов с разной исходной концентрацией данного вещества. Этот метод называют иногда методом Оствальда – Нойеса; он особенно удобен для определения порядка реакций, протекающих между газообразными веществами.
2. Метод подбора уравнений, основанный на подстановке экспериментальных данных по концентрации вещества для каждого момента времени в кинетические уравнения реакций различных порядков. Определяемый порядок реакции соответствует тому уравнению, для которого при различных начальных концентрациях вещества и в различные моменты времени при заданной Т константа скорости будет оставаться постоянной.
3.
Графический
метод,
основанный на определении такой функции
концентрации, которая на графике
зависимости ее от времени дает прямую
линию. Для реакций 1-го порядка такой
функцией является ln
(a–x),
для реакций 2-го порядка –
![]() (если a
= b),
для реакций 3-го порядка –
(если a
= b),
для реакций 3-го порядка –
![]() ,
что вытекает из полученных ранее
кинетических уравнений. По наклону
полученной прямой на таком графике
вычисляют константу скорости, как это
видно из рис. 4.
,
что вытекает из полученных ранее
кинетических уравнений. По наклону
полученной прямой на таком графике
вычисляют константу скорости, как это
видно из рис. 4.
Дифференциальные методы основаны на использовании уравнения для скорости реакции в его дифференциальной форме.
1. Метод Вант-Гоффа. Проводят реакцию так, что берут все вещества, кроме одного (скажем, А) в избытке. Реакцию проводят дважды, задаваясь разными начальными значениями концентрации. Тогда можно записать два уравнения скорости
–
![]() =
k1
=
k1
![]() ; –
; –
![]() = k1
= k1
![]() .
.
Разделим уравнения друг на друга и прологарифмируем:
1-й порядок |
2-й порядок (a = b) |
|
|
ln (a – x) = ln a – kt |
=
|
|
|
3-й порядок |
2-й порядок (a b) |
|
|
|
kt
(a
– b)
=
= k (a – b) t – . |
Рис. 4. Графический метод определения порядка химической реакции |
|
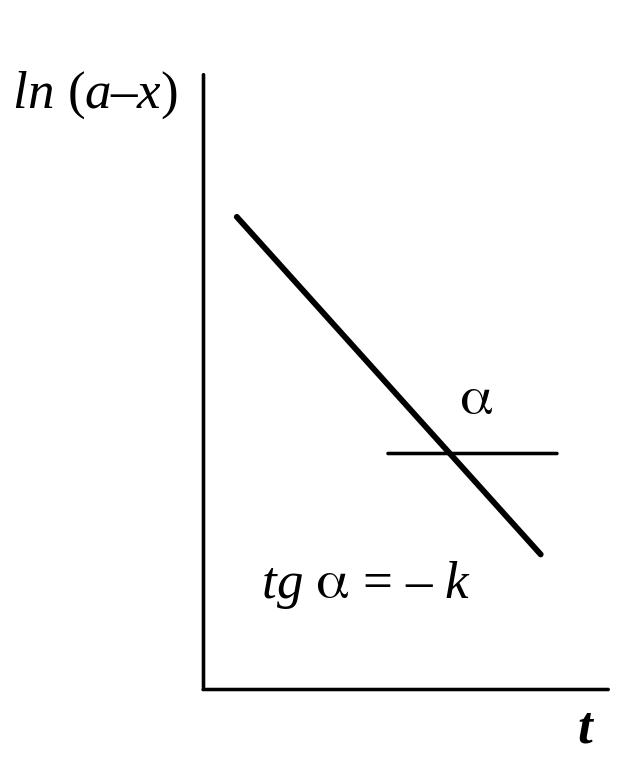
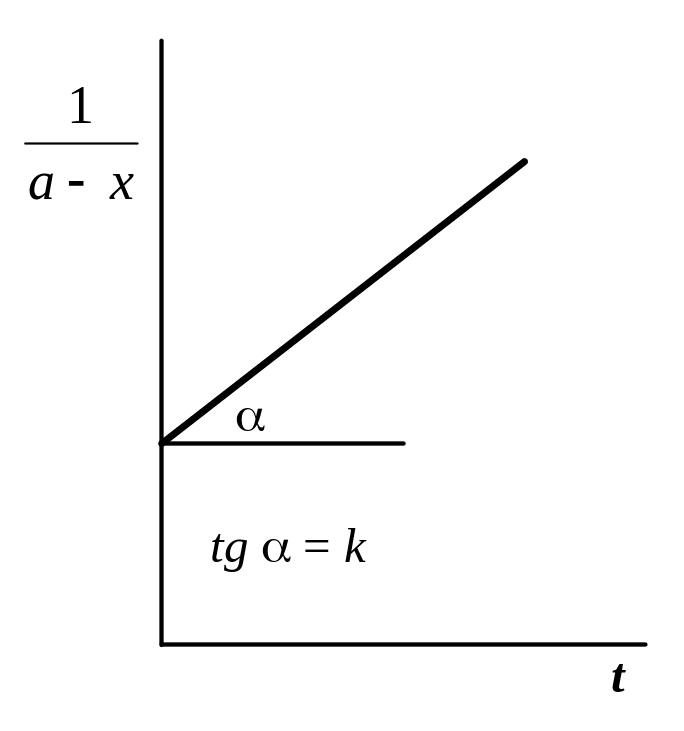
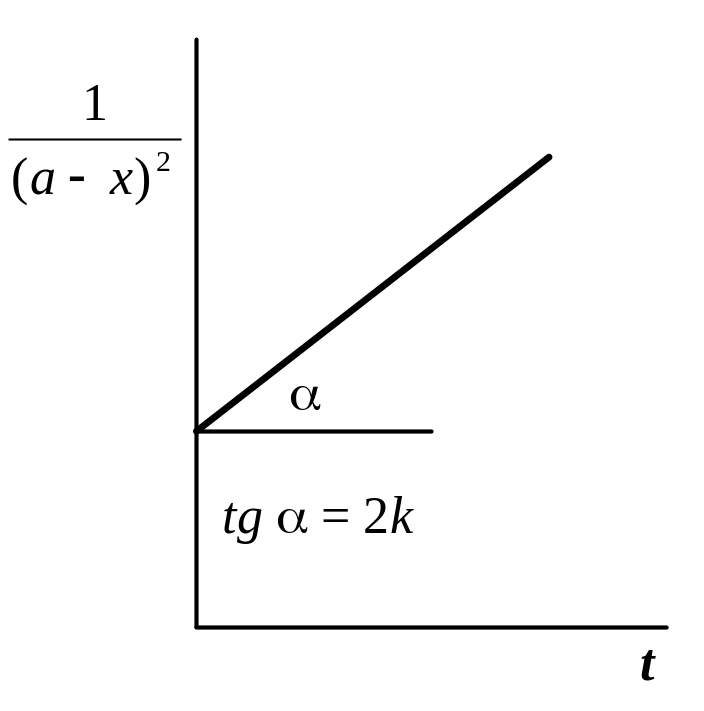
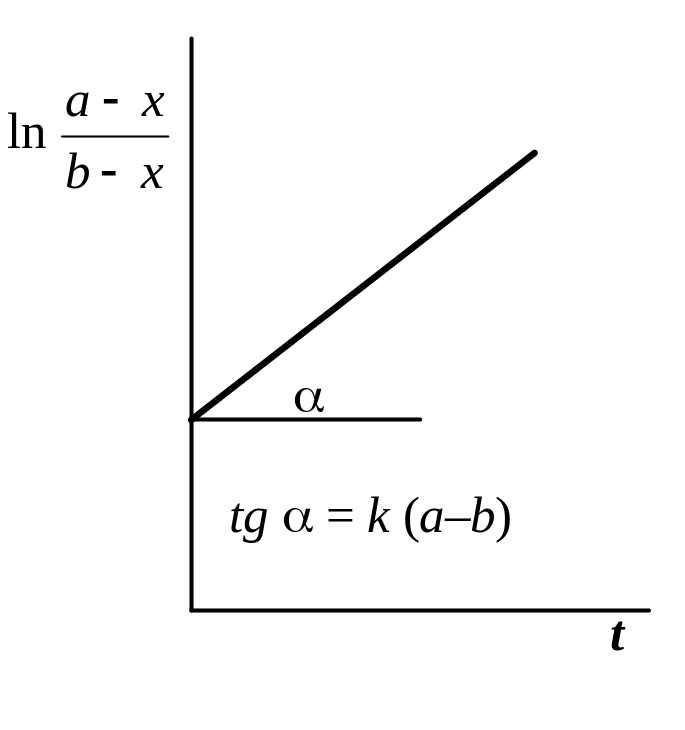
![]() =
=
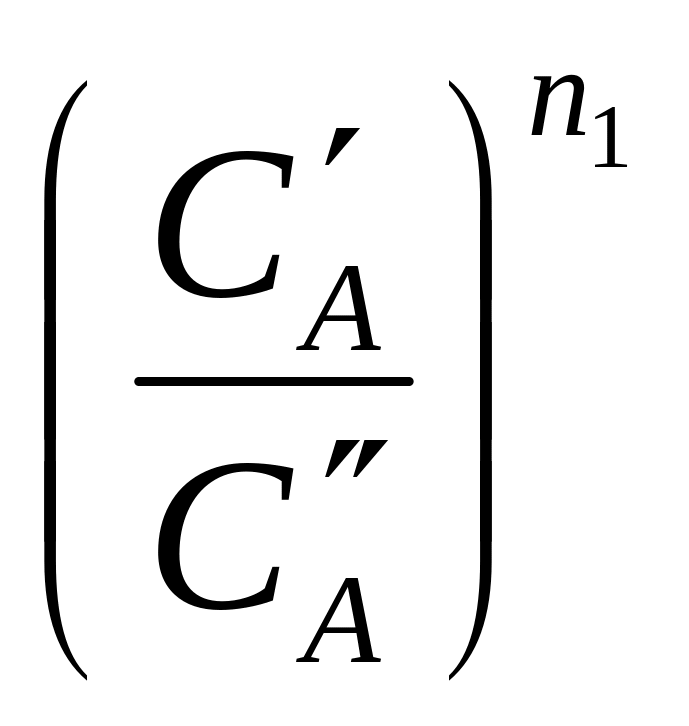 ;
;
lg (CA /t) – lg (CA /t) = n1 (lg CA – lg CA) ;
n1
=
![]() .
.
Дифференциалы заменяем конечными разностями
n1
=
![]() .
.
Это уравнение и позволяет определить порядок реакции по веществу А. Для этого надо определить изменение концентраций CA и CA за время t , задавая разные исходные концентрации вещества А, а концентрации всех остальных веществ выбирая очень большими. Значения CA и CA , входящие в знаменатель, нужно брать средними для данного промежутка времени. Аналогично определяют порядки реакций по другим веществам.
2. Графические варианты метода Вант-Гоффа основаны на использовании уравнения
v = kC n ,
которое можно преобразовать
lg v = lg k + n lgC .
На графике в координатах lg v – lgC зависимость изображается прямой линией, угловой коэффициент которой равен n. Скорость определяется тангенсом угла наклона касательной к кривой С = f (t). В зависимости от того, какая определяется скорость – в начальный момент реакции или в различные промежутки времени от начала ее – различаются два графических варианта этого метода.
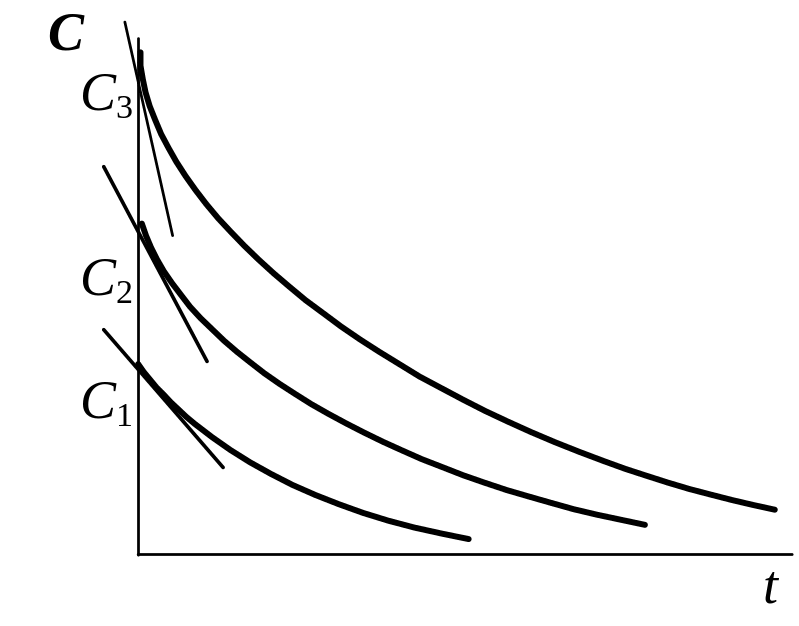
Вариант А. Определяют начальные скорости реакции при различных исходных концентрациях вещества. Для этого проводят касательную к точке кривой, соответствующей началу реакции. Логарифмы начальных скоростей наносят на график lg v = f (lgC). Наклон прямой соответствует порядку реакции nc , который называют концентрационным, или истинным (см. рис. 5). При таком определении порядка реакции образующиеся промежуточные продукты не могут влиять на ее скорость.
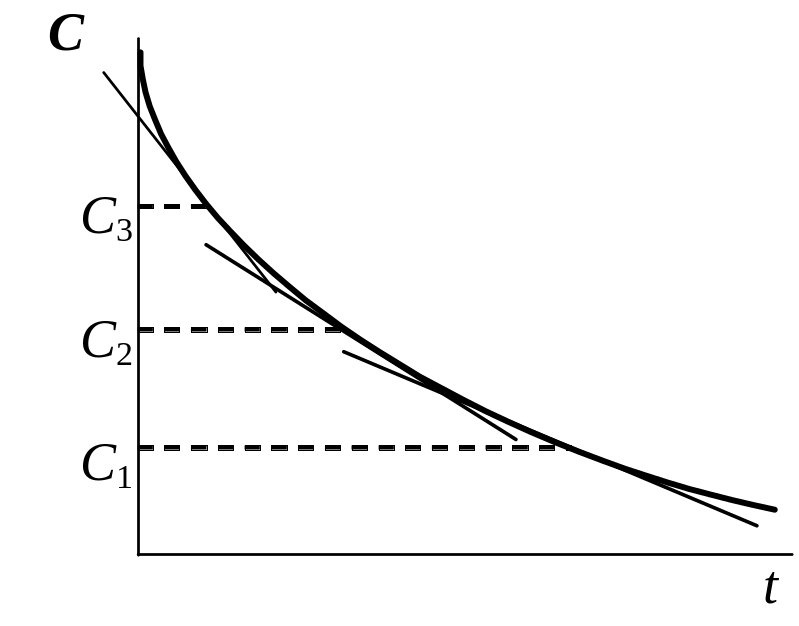
Вариант Б. Определяют скорость реакции в различные моменты времени от начала реакции, используя данные одного опыта. Для этого измеряют тангенс угла наклона касательных к кривой С = f (t) при различных значениях t. Затем строят зависимость lg v = f (lgC) , из которой находят порядок реакции, называемый временным (nt ) (рис. 6).
|
|
Рис. 5. Экспериментальное определение концентрационного (истинного) порядка реакции |
|
|
|
Рис. 6. Определение временного порядка реакции |
|
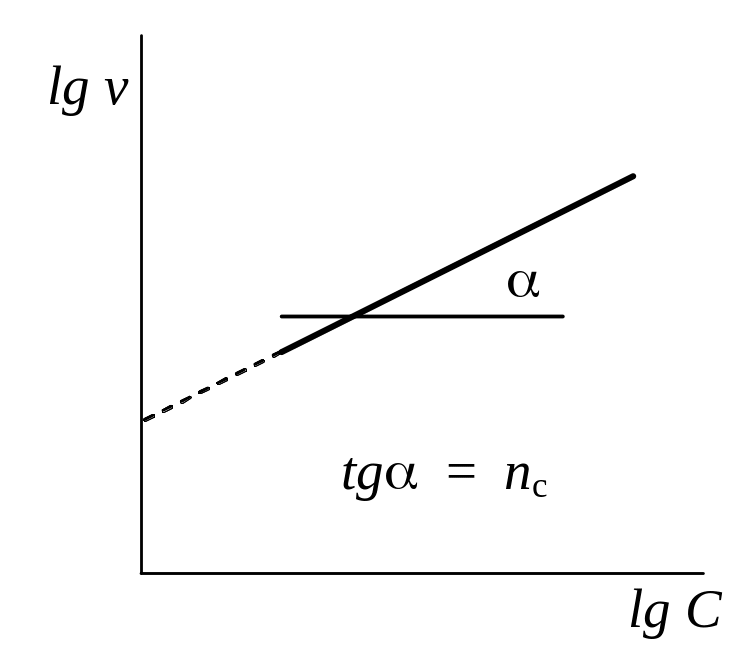
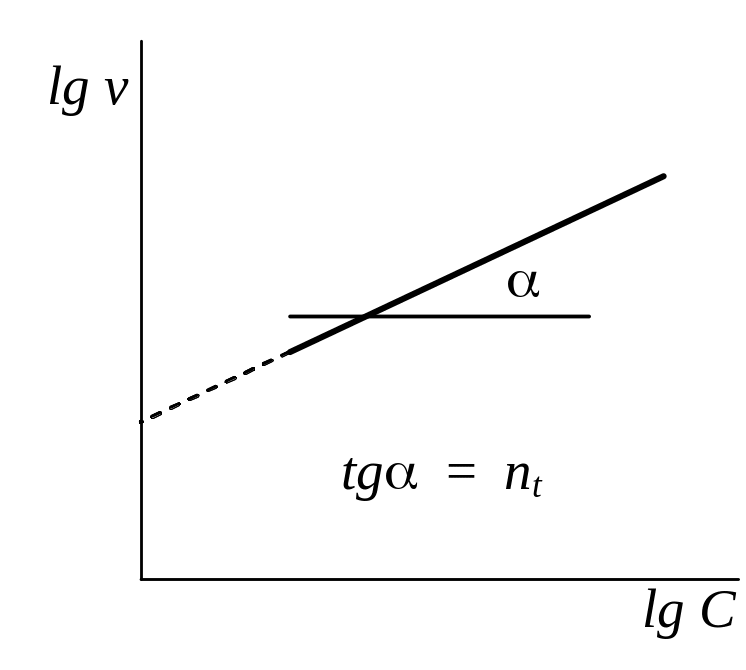
Данным способом можно обнаружить влияние промежуточных продуктов реакции на ее скорость: если промежуточные продукты искажают ход реакции, то временной и концентрационный порядки реакции могут быть разными. Кроме того, указанием на то, что реакция протекает через различные промежуточные стадии, является дробный порядок реакции.
Таким образом, необходимыми экспериментальными данными для определения порядка реакции являются данные об изменении концентрации реагирующих веществ во времени; такие данные надо получить для разных исходных концентраций веществ.