

Распределение ионов в растворе
В теории электролитов очень важным является вопрос о распределении ионов в растворе. По первоначальной теории электролитической диссоциации, основанной на физической теории растворов Вант-Гоффа, считалось, что ионы в растворах находятся в состоянии беспорядочного движения в состоянии, подобном газообразному.
Однако представление о беспорядочном распределении ионов в растворе не соответствует действительности, так как онo не учитывает электростатического взаимодействия ионов. Электрические силы проявляются на относительно больших расстояниях, и в сильных электролитах, где диссоциация велика, а концентрация ионов значительна и расстояния между ними невелики, электростатическое взаимодействие ионов настолько сильно, что оно не может не сказываться на характере их распределения. Возникает тенденция к упорядоченному распределению, аналогичному распределению ионов в ионных кристаллах, где каждый ион окружён ионами противоположного знака.
Распределение ионов будет определяться соотношением электростатической энергии и энергии хаотического движения ионов. Эти энергии сравнимы по величине, поэтому реальное распределение ионов в электролите является промежуточным между беспорядочным и упорядоченным. Около каждого иона образуется своеобразная ионная атмосфера, в которой преобладают ионы противоположного (по сравнению с центральным ионом) знака. В этом заключается своеобразие электролитов и трудности, возникающие при создании теории электролитов.
Лекция 49
Теория электролитов Дебая и Гюккеля
Основные положения современной теории растворов электролитов были сформулированы в 1923 г. Дебаем и Гюккелем. Для статистической теории электролитов исходным является следующее положение: ионы распределены в объеме раствора не хаотически, а в соответствии с законом кулоновского взаимодействия. Вокруг каждого отдельного иона существует ионная атмосфера (ионное облако) сфера, состоящая из ионов противоположного знака. Ионы, входящие в состав сферы, непрерывно обмениваются местами с другими ионами. Все ионы раствора равноценны, каждый из них окружен ионной атмосферой, и в то же время каждый центральный ион входит в состав ионной атмосферы какого-либо другого иона. Существование ионных атмосфер и есть тот характерный признак, который, по Дебаю и Гюккелю, отличает реальные растворы электролитов от идеальных.
Энергия, связанная с ионной атмосферой, имеет электрическое происхождение, поэтому она должна быть функцией плотности электрического заряда и потенциала, создаваемого ионной атмосферой. Тепловое движение ионов в ионной атмосфере приводит к тому, что дискретные заряды этих ионов как бы размазываются. В результате ионную атмосферу, состоящую из отдельных ионов, в среднем за некоторый промежуток времени можно моделировать облаком размазанного заряда, плотность которого уменьшается по мере удаления от центрального иона. Для связи между средней плотностью заряда и отвечающим ему средним значением потенциала можно использовать уравнение Пуассона
2
= –
![]() ,
,
где
2
– оператор Лапласа 2
=
![]() +
+
![]() +
+
![]() .
.
Поскольку ионная атмосфера обладает шаровой симметрией, то уравнение Пуассона, записанное в сферической системе координат, принимает вид
![]() +
+
![]() = –
= –
и связывает объемную плотность заряда с потенциалом на расстоянии r от центрального иона. В уравнение Пуассона входят две неизвестные величины: и . Для их определения необходимо иметь второе уравнение, связывающее обе эти переменные. Дебай и Гюккель получили нужное уравнение следующим образом.
Для раствора в целом и для любого заданного его объема, достаточно большого по сравнению с размерами иона, справедливо условие электронейтральности
ni ezi = 0 ,
где ni – число ионов i-го сорта в единице объема (если выбран объем раствора, равный единице). Однако заряд некоторого элемента объема dV, находящегося вблизи какого-либо иона, будет отличаться от нуля вследствие существования ионной атмосферы. Если центральный ион, расположенный в начале координат, заряжен положительно, то элемент объема dV будет обладать избыточным отрицательным зарядом. Предполагая, что к распределению ионов в растворе применим принцип Больцмана и что силы, действующие между ионами, по своей природе электростатические, число отрицательных и число положительных ионов в элементарном объеме dV можно выразить как
dn–
= n–
![]() dV
, dn+
= n+
dV
, dn+
= n+![]() dV
.
dV
.
Заряд элемента объема будет равен
dV
= ez–
n–
exp
![]() dV
– ez+
n+
exp
dV
– ez+
n+
exp
![]() dV
,
dV
,
а плотность заряда
= ez– n– exp – ez+ n+ exp =
=
e
zi
ni
exp
![]()
(учитывая, что в растворе находятся различные ионы, и приписывая zi знак, отвечающий заряду иона). Подстановка из этого выражения в уравнение Пуассона дает уравнение, которое в общем виде нельзя проинтегрировать. Чтобы упростить его, Дебай и Гюккель предположили, что ezi kT , то есть что электростатическое взаимодействие мало по сравнению с термической энергией. Разложив показательную функцию в ряд
exp
= 1 –
![]() +
+
![]() 2
+ … ,
2
+ … ,
они ограничились только двумя его первыми членами, получив для следующее уравнение:
=
e
zi
ni
–
![]()
zi2
ni
= –
zi2
ni
zi2
ni
= –
zi2
ni
(так как первый член правой части уравнения по условию электронейтральности равен нулю). Если ввести обозначение
=
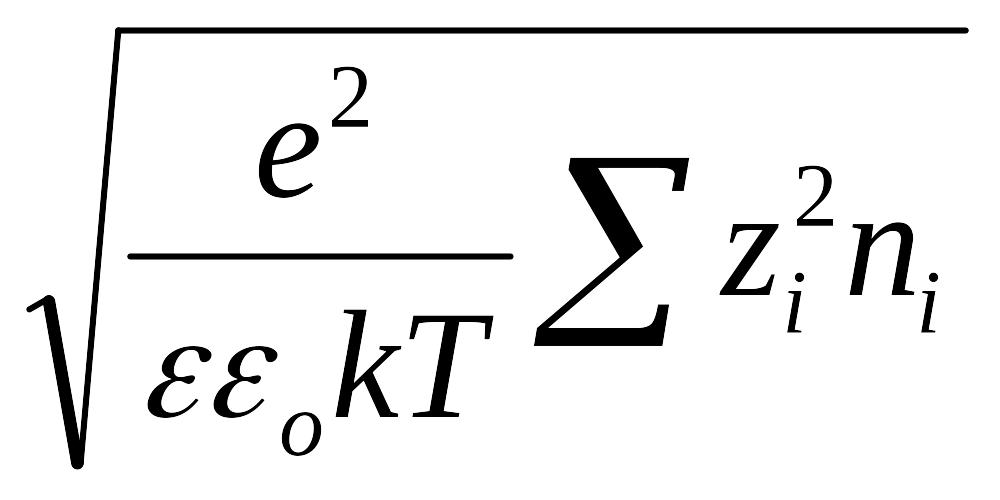 ,
,
то после подстановки выражения для в уравнение Пуассона получаем дифференциальное уравнение вида
+ = 2 .
Общее решение этого уравнения второго порядка имеет вид
=
А1
![]() + А2
+ А2
![]() .
.
Константы интегрирования А1 и А2 находят из пограничных условий. Как следует из основных законов электростатики, при r 0; это условие выполняется в том случае, если А2 = 0, так как иначе с удалением от центрального иона будет стремиться к бесконечности. Для определения А1 в первом приближении теории Дебая – Гюккеля предполагается, что ионы можно отождествить с материальными точками, обладающими определенным зарядом. В этом случае при r 0 должен стремиться к потенциалу самого иона i , то есть
r
0
= i
=
![]() ,
,
что является вторым пограничным условием. Если е–r разложить в ряд, то получим
=
А1
= А1
![]() (1 – r
+
(r)2
– …) .
(1 – r
+
(r)2
– …) .
При r 0 всеми членами ряда можно пренебречь по сравнению с 1. Тогда
r
0
i
= А1/r
, A1
=
![]() ,
=
exp (–r)
.
,
=
exp (–r)
.
Величина в последнем уравнении представляет собой среднее значение потенциала в точке r, создаваемое ионной атмосферой и центральным ионом. Для вычисления ион-ионного взаимодействия представляет интерес не общий потенциал , а та его часть а , которая создается ионной атмосферой в месте расположения центрального иона. а называется потенциалом ионной атмосферы. Его находят по правилу суперпозиции (то есть наложения) потенциалов
а = – i = (exp (–r) – 1) .
Для вычисления энергии взаимодействия необходимо определить потенциал ионной атмосферы в точке нахождения центрального иона, то есть найти предел а при r 0. Это можно сделать, используя тот же прием – разлагая показательную функцию в ряд и пренебрегая высшими членами разложения:
lim а = = – .
Величину можно рассматривать как потенциал, создаваемый в точке нахождения центрального иона другим ионом с противоположным знаком, находящимся от центрального иона на расстоянии 1/. Величина 1/ называется характеристической длиной. Так как потенциал создается не единичным ионом, а всей ионной атмосферой, то 1/ можно отождествить с радиусом ионной атмосферы. Величину можно рассчитать по вышеприведенному уравнению.
В первом приближении теории Дебая и Гюккеля предполагается, что энергия межионного взаимодействия имеет электростатическое происхождение. Энергия взаимодействия ионов может быть определена поэтому как энергия заряжения центрального иона в электрическом поле ионной атмосферы. Известно, что энергия заряжения частицы от 0 до заряда q в поле равна
![]() =
=
![]()
![]() =
=
![]() =
q
=
ezi
(–
)
=
=
q
=
ezi
(–
)
=
=
–
![]()
(вместо q подставили заряд центрального иона, а вместо – величину ).
Найдем выражение для коэффициента активности электролита. Коэффициенты активности первоначально были введены как эмпирические величины; необходимость их введения в уравнения для идеальных систем обусловлена различием между реальными и идеальными растворами. Коэффициенты активности не зависят от метода их определения и при заданных условиях и составе будут иметь одно и то же значение для любых типов равновесия. Таблицы, в которых приведены эмпирические коэффициенты активности, представляют большую ценность. В то же время в пределах теории Льюиса коэффициенты активности оказываются лишь формальными поправочными множителями, не связанными непосредственно с природой растворов и не поддающимися теоретическим расчетам.
Реальные растворы отличаются от идеальных энергией взаимодействия образующими их частиц. Сопоставление выражений для химических потенциалов частиц в реальных растворах
i = io + RT ln Ni + RT ln i
и в идеальных растворах
![]() =
io
+ RT
ln
Ni
=
io
+ RT
ln
Ni
показывает, что
RT ln i = i – = i .
Таким образом, коэффициент активности отражает изменение энергетического состояния ионов, обусловленное межионным (межчастичным) взаимодействием. Расчет взаимодействия, а следовательно, и коэффициента активности возможен, если известны структура раствора и природа сил взаимодействия между образующими его частицами. Ни то, ни другое не известно с достаточной степенью точности, поэтому для решения проблемы приходится прибегать к некоторым предположениям и создавать модели растворов электролитов.
По модели Дебая и Гюккеля, энергия взаимодействия иона с ионной атмосферой, равная энергии заряжения, определяется выражением
i = – NA .
Для коэффициента активности
ln
i
= – NA
![]()
.
.
Подстановка значения приводит к
ln
i
= – NA

![]() ,
,
где I – ионная сила раствора, определяемая соотношением
I = сi zi2 .
(![]() =
=
![]() =
=
![]() =
=
= ;
ni = ион /м 3; ni / NA = моль / м 3; ni /(1000NA) = ci = моль/л ).
После перехода к десятичному логарифму и подстановки численных значений постоянных величин
lg
i
= – 1,8246106
(T)–
3/2 zi2![]() .
.
Для данного растворителя и определенной Т можно написать
lg i = – zi2 h .
Для среднего ионного коэффициента активности, который в отличие от коэффициента активности отдельного вида ионов определяется экспериментально, справедливо уравнение
lg = – z+ z–h . (1)
Для водных растворов при 25оС, допуская равенство диэлектрических проницаемостей раствора и растворителя (78,54), h = 0,51. Для 1-1 зарядных электролитов
lg
= – h![]() = – 0,51
.
= – 0,51
.