

9.6. Нарушение кислотно-основного баланса
Нарушение кислотно-основного баланса (КОБ) может осложнять течение многих заболеваний, являясь следствием изменений газового состава крови, метаболических расстройств, которые возникают, например, при недостаточности дыхания, кровообращения, печени, почек, при эндокринных заболеваниях, патологии желудочно-кишечного тракта, системы крови и др.
Для оценки характера изменений КОБ принято оценивать концентрацию Н+ в артериальной крови, т.е. определять рН крови; рН — отрицательный логарифм концентрации водородных ионов. Увеличение рН крови более 7,45 свидетельствует о защелачивании (алкалемии), уменьшение рН менее 7,55 — о закислении (ацидемии).
При существенных сдвигах рН в ту или иную сторону нарушаются функции клеток, прежде всего работа их многочисленных ферментных систем, изменяются направленность и интенсивность окислительно-вос- становительных процессов, способность гемоглобина связывать и отдавать кислород. Изменяется водный-электролитный баланс, увеличивается проницаемость клеточных мембран и др.
Уменьшение рН крови менее 6,8 и увеличение более 7,7 несовместимы с жизнью. Для поддержания концентрации ионов водорода и соответственно рН в таком узком диапазоне в организме существуют специальные системы — это буферные системы крови и клеток и физиологические — главным образом легкие и почки.
9.6.1. Основы регуляции кислотно-основного баланса
Роль буферных систем. Буфер — это слабая кислота или основание, которые противостоят изменению рН при добавлении сильной кислоты или основания. Буферные системы клеток и плазмы крови играют первостепенную роль в поддержании относительно узкого диапазона рН, в котором протекают физиологические клеточные и внеклеточные процессы. Главной буферной системой крови служит система С02—бикарбонат, которая действует во внеклеточной и внутриклеточной.жидкостях организма. Кроме того, внутриклеточные белки, гемоглобин, белки плазмы и составные элементы костей (например, карбонаты и коллаген) также играют роль буферов.
Анион бикарбоната присутствует в большинстве жидкостей организма и является его главным щелочным резервом. Бикарбонат реагирует с ионом водорода, образуя угольную кислоту, которая существует в равновесии с С02 (Н+ + НС03~ о Н2С03 <-> С02 + Н20). Превращение Н2С03 в Н20 и С02 катализируется ферментом карбоангидразой. В ходе метаболизма образующиеся Н+ взаимодействуют с бикарбонатом, образуя Н2С03, а затем С02 и Н20. Углекислый газ выводится легкими. И, наоборот, когда С02 образуется в процессе клеточного метаболизма, угольная кислота диссоциирует на Н+ и НС03~.
рН системы, в которой протекают эти реакции, рассчитывается на основе уравнения Гендерсона-Хассельбаха. Оно выводится из уравнения для константы диссоциации (Ка) угольной кислоты:
_ [Н+]х[НС03] а [Н2С03] '
или в логарифмической форме:
lg[H+] + 1д[НС03-]
|дка =
[Н2С03]
Поскольку рН — это отрицательный логарифм [Н+], уравнение можно записать так: [НСО "]
рН = рКа + |д( 0>03хр^02 ).
Величина рКа для буферной системы С02—бикарбонат равна 6,1 Концентрация бикарбоната в плазме артериальной крови здорового человека — 24 ммоль/л, а нормальное парциальное давление углекислого газа — 40 мм рт.ст. Следовательно, в норме рН артериальной крови составляет 7,4:
6,1 + Ig24 ф 0,03x24
Величина рКа для буферов-белков равна 7,4 (Н-белок <-> Н+ + белок). Величина рКа для буфера-фосфата — 6,8 (Н2Р04 <-> Н+ + НР04 2) Белки и фосфаты являются главными внутриклеточными буферами.
Изменение функций внешнего дыхания. Вдыхаемый воздух содержит незначительное количество С02. Почти вся углекислота крови является продуктом клеточного метаболизма. По мере образования в процессе клеточного метаболизма С02 легко диффундирует в капилляры и транспортируется к легким в трех основных формах:
растворенная С02;
анион бикарбоната;
карбаминовое соединение (рис. 9.7).
С02 очень хорошо растворяется в плазме. Количество растворенной в плазме С02 определяется произведением ее парциального давления и коэффициента растворимости. Около 5 % общей двуокиси углерода в артериальной крови находится в форме растворенного газа, а 90 % — в форме бикарбоната. Последний является продуктом реакции С02 с водой с образованием Н2С03 и ее диссоциацией на водород и ион бикарбоната: С02 + Н20 о Н2С03 нН+ + НС03~ Реакция между С02 и Н20 протекает медленно в плазме и очень быстро в эритроцитах, где присутствует внутриклеточный фермент карбоангидраза. Она облегчает реакцию между С02 и Н20 с образованием Н2С03; вторая реакция протекает очень быстро без катализатора.
По мере накопления НС03~ внутри эритроцита анион диффундирует через клеточную мембрану в плазму. Мембрана эритроцита плохо проницаема для Н+, как и вообще для катионов, поэтому ионы водорода остаются внутри клетки. Электрическая нейтральность клетки в процессе диффузии НС03~ в плазму обеспечивается потоком ионов хлора из плазмы в эритроцит, что формирует так называемый хлоридный сдвиг.
Часть Н+, остающихся в эритроцитах, соединяется с гемоглобином. В периферических тканях, где концентрация С02 высока и значительное количество Н+ накапливается эритроцитами, связывание Н+ облегчается
Рис.
9.7.
С02-транспорт
в крови, иллюстрирующий образование
НС03~
и карбаминовых соединений, хлоридный
сдвиг и связывание Н+.
При поглощении 02
и высвобождении С02
в легочных капиллярах представленные
реакции протекают в обратном порядке
(поМайклА Гриппи, 1997)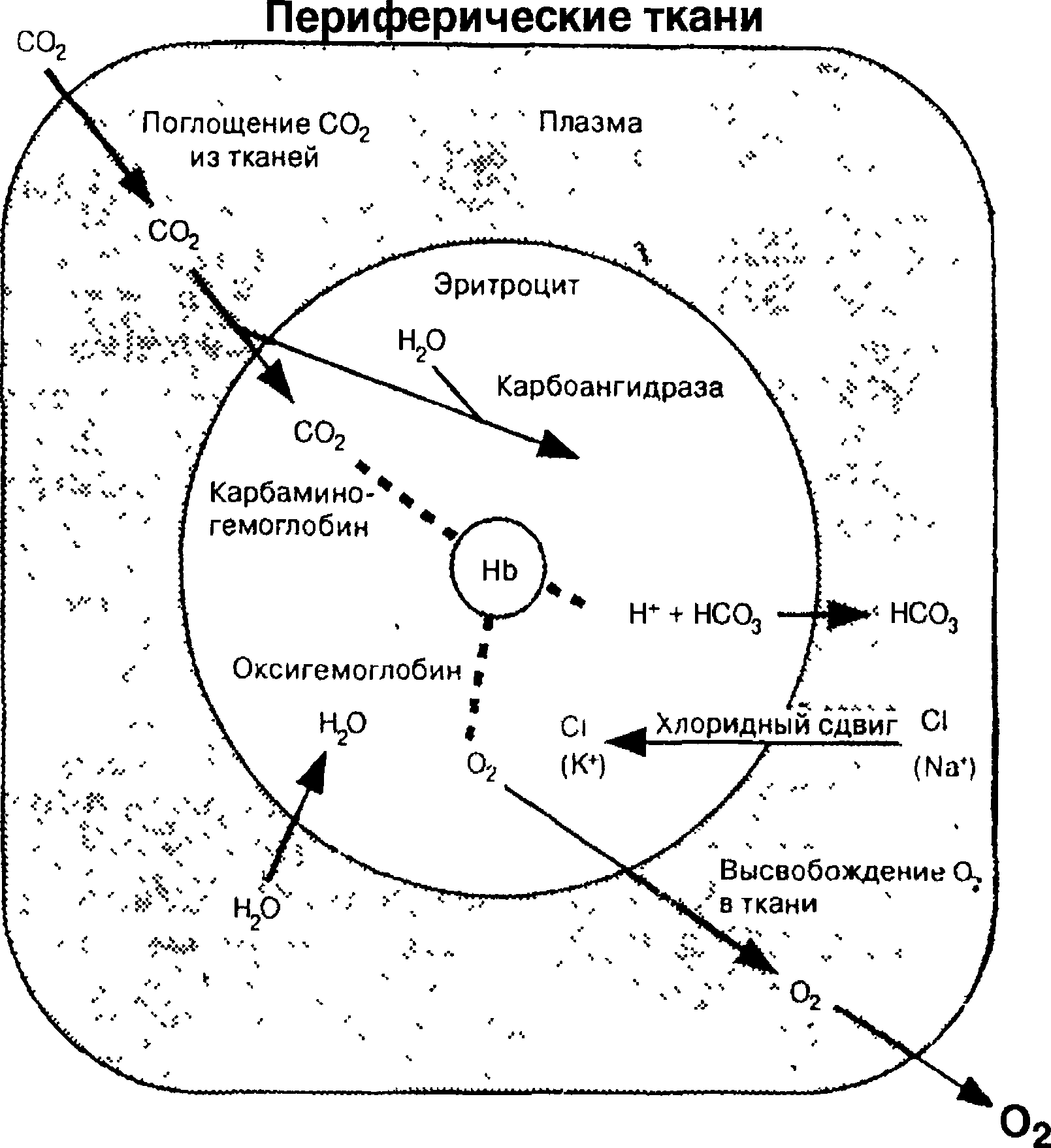
деоксигенацией гемоглобина. Восстановленный гемоглобин лучше связывается с протонами, чем оксигенированный. Таким образом, деокси- генация артериальной крови в периферических тканях способствует связыванию Н+ посредством образования восстановленного гемоглобина. Это увеличение связывания С02 с гемоглобином известно как эффект Холдейна.
Третьей формой транспорта С02 являются карбаминовые соединения, образованные в реакции С02 с концевыми аминогруппами белков крови. Основным белком крови, связывающим С02является гемоглобин. Этот процесс описывается реакцией: Hb-NH2 + С02 Hb-NH х СООН Hb-NHCOO" + Н+. Реакция С02 с аминогруппами протекает быстро. Как и в случае более легкого связывания С02 с восстановленным гемоглобином, образование карбаминовых соединений легче протекает с деокси- генированной формой гемоглобина. Карбаминовые соединения составляют около 5 % общего количества С02, транспортируемого артериальной кровью.
Регуляция выделения С02 достигается изменением скорости объема легочной вентиляции, т.е. зависит от величины минутной альвеолярной вентиляции (МАВ). Повышение МАВ приводит к снижению артериального рС02 и наоборот. Афферентные сигналы, изменяющие минутную альвеолярную вентиляцию, связаны с хеморецепторами, которые регулируют функции дыхательного центра. Эти рецепторы находятся в продолговатом мозге, аортальном и каротидном тельцах и реагируют на изменения рС02 и концентрации Н+.
Легкие — это первая линия защиты в поддержании кислотно-основного гомеостаза, поскольку они обеспечивают механизм почти немедленной регуляции выделения кислоты. В то же время любые нарушения дыхания, сопровождающиеся увеличением или уменьшением минутной альвеолярной вентиляции, могут стать причиной развития нарушений КОБ.
Роль почек. Количество нелетучих кислот, образующихся в процессе метаболизма белков и других веществ, гораздо меньше, чем летучих. Тем не менее, почки выделяют от 50 до 100 ммоль нелетучих1 кислот в сутки. Их выделение происходит в проксимальных канальцах и собирательных трубках почек, где секретируются протоны, а в качестве буферных систем участвуют фосфаты, сульфаты (т.е. титруемые кислоты) и аммиак. Однако до того как может произойти экскреция всех кислот, почки должны реабсорбировать НС03~, профильтровавшийся в клубочках.
Способность канальцев почек к реабсорбции НС03~ высока. В среднем человек выделяет менее 5 ммоль НС03" в сутки. Самьм важным местом реабсорбции НС03~ являются проксимальные канальцы, где посредством специального механизма происходит всасывание 90 % бикарбоната. Угольная кислота образуется в клетке из воды и С02 под действием карбоангидразы, Н+ активно переносятся через люминальную мембрану Na+-, Н+-обменником (рис. 9.8). Затем НС03~ транспортируется через базолатеральную мембрану. Секретируемый Н+ быстро соединяется с фильтруемым НС03~, образуя угольную кислоту (Н2С03). Угольная кислота превращается в воду и углекислый газ с помощью карбоангидразы (КА) на люминальной стороне щеточной каемки проксимального канальца. С02 диффундирует обратно в клетку проксимального канальца, где соединяется с Н20 и образует угольную кислоту, завершая тем самым этот цикл (рис. 9.8).
Некарбоновые кислоты секретируются вставочными клетками собирательных трубок коры и наружного мозгового слоя почек. Секрецию Н+ в просвет канальцев осуществляет Н+-АТФаза, тогда как в реабсорбции НС03~ через базолатеральную мембрану участвует обменник С1~, НС03~ (рис. 9.9).
Главным фактором, от которого зависит количество выделяемых кислот, является присутствие буферов в моче. Максимальный рН жидкос
-
Базолатеральная Просвет Базолатеральная Просвет
KA
—
карбоангидраза. (по Френк К. Брозиус,
1997).
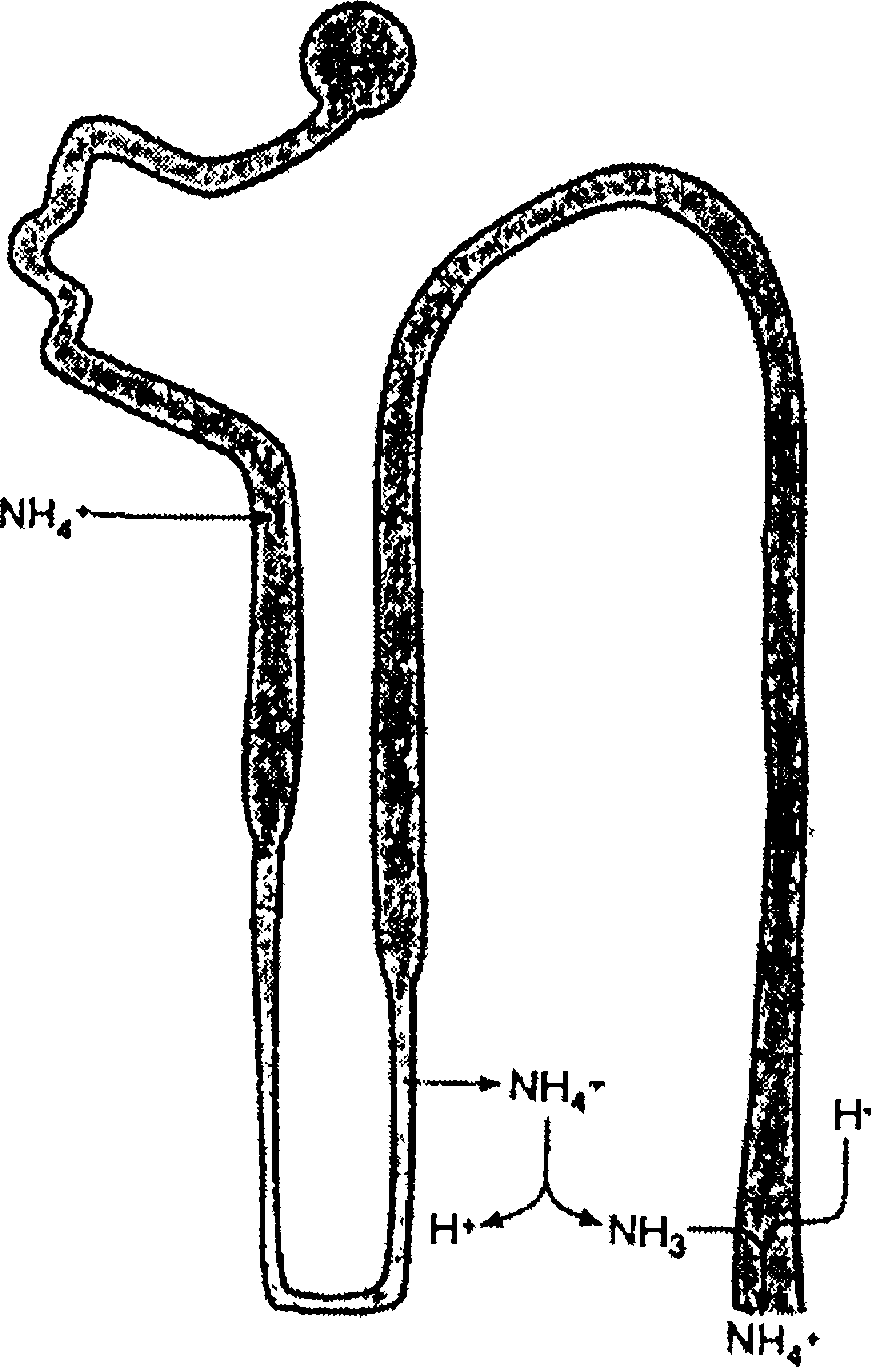
Рис.
9.8.
Реасорбция бикарбоната Рис.
9.9.
Секреция Н+
вставочными
в клеткахпроксимального канальца. а-клетками собирательной трубки.
АДФ — аденозиндифосфат; АТФ — аденозинтрифосфат. (по Френк К. Брозиус, 1997).
Рис. 9.10. Транспорт NH3 и NH4 в почке.
NH4+ образуется и секретируется клетками проксимального канальца, а затем реабсор- бируется в восходящем отделе петли Генле и концентрируется в мозговом слое почки. Небольшое количество NH4+ диссоциирует на NH3 и Н+, последний реабсорбируется. NH3 может диффундировать в собирательную трубку, где служит буфером для ионов Н\ секретируемых вставочными клетками.
ти в просвете собирательной трубки — 4,0 (Н+= 0,1 моль/л). Поэтому только 0,1—0,2% суточной нагрузки кислот (50—100 ммоль) могут быть выведены в форме незабуференных ионов Н+. Остальная часть Н+ в моче должна быть выведена в форме буферов, обычно таких, как фосфаты и аммоний. Концентрация аммония регулируется преимущественно почками и колеблется в зависимости от КОБ (рис. 9.10). Объем суточной секреции кислот в наибольшей степени зависит от количества выделяемого аммония.
|
Факторами,
регулирующими транспорт Н+
и НС03~
в проксимальных канальцах почек,
являются: рС02,
фильтруемая нагрузка НС03~,
карбоан- гидраза, паратиреоидный
гормон, концентрация К+
и НР04~2
в сыворотке. В собирательных трубках
регуляция транспорта катионов и
бикарбоната обеспечивается: градиентом
рН, разностью электрических потенциалов |
|
|
юз |
|
|
Велик |
|
|
16 |
|
|
|
|
|
|
|
|
OA ■ v ' V |
|

CI
-
103
Na
'
142