

СюТоксическое действие
Расслабление стенок кровеносных сосудов
Антимикробное действие
Детоксикация Н20
28
LOOH
НООН
нею
L0
НО-
НО
Fe2+-
JW
10
^Токсическое действие
Рис. 2.6. Метаболизм супероксидного радикала.
Объяснения в тексте.
лентности супероксидные радикалы превращаются в перекись водорода; эта реакция катализируется ферментом супероксиддисмутазой (реакция 2):
2-02" Н202 + 02
Клетки-фагоциты используют перекись водорода, превращая ее в гипохлорит — соединение, разрушающее стенки бактериальных клеток; эта реакция катализируется ферментом миелопероксидазой (реакция 3):
Н202 + С|--»Н20 + СЮ-.
Избыток перекиси водорода удаляется под действием двух ферментов: глутатион-пероксидазы или каталазы (4 на рис. 2.6):
Н202 + 2GSH (глутатион) -> Глутатионпероксидаза 2Н20 + GSSG;
2Н202 Каталаза 2Н20 + 02.
Радикал гидроксила. В условиях патологии могут произойти нарушения либо системы защитных ферментов (в частности, снижение активности СОД), либо ферментных систем, связывающих ионы железа в плазме крови (церулоплазмин и трансферрин) и в клетках (ферритин). В этом случае супероксидные радикалы и перекись водорода вступают в альтернативные реакции:
образование двухвалентного железа из трехвалентного (рис. 2.6, 7):
Fe3+ + ю2- Fe2+ + 02;
реакция перекиси водорода и гипохлорита с ионами двухвалентного железа (рис. 2.6, 9 и 10):
Fe2+ + Н202 Fe3+ + НО" + НО* (реакция Фентон); Fe2+ + СЮ" + Н+ Fe3+ + CI" + НО- (реакция Осипова)
.
Совокупность продуктов, образуемых активированными клетками- фагоцитами (радикалы супероксида и гидроксила, перекись водорода и гипохлорит), называют активными формами кислорода; некоторые авторы называют гипохлорит и продукты его метаболизма в тканях (такие, как хлорамины R-NHCI), активными формами хлора.
Радикалы гидроксила химически исключительно активны и вызывают повреждение белков, нуклеиновых кислот и липидов биологических мембран. Особенно тяжелые последствия имеют две последние реакции. Радикалы ЮН вызывают разрыв нитей ДНК, оказывают в зависимости от ситуации, мутагенное, канцерогенное или цитостатическое действие. Вместе с тем, реагируя с ненасыщенными жирными кислотами, входящими в состав мембранных липидов, радикалы гидроксила инициируют цепную реакцию их пероксидации (перекисного окисления).
Цепное
окисление липидов.
Реакция цепного окисления липидов
играет исключительную роль в клеточной
патологии. Она протекает в несколько
стадий, которые получили название
инициирование, продолжение,
разветвление и обрыв цепи (рис. 2.7).
Рассмотрим эти стадии под
lh
^ loo
HOH
робнее.
QO
Jl
оо
ж
QO
lh
lh
но-
->•
loo*
?
ноон
ноон
Звено
цепи
и
Первичная цепная реакция
lh
I
00
l*
оо
—i-
OO
,-i-
lh
I
ноон
lx>
loo
*
Fev
re*"1
tmct,+ f гс
*Л
Г Li
I
'
с П
«
*
I
00
^
T ^
i
lh -
ar ▼
»
iL'
§
ОО ~»4
X j
loo
*
loh
loh
loo*
Рис. 2.7. Реакция цепного окисления липидов.
А — реакция с неразветвленной цепью, Б — разветвленная цепная реакция
Инициирование цепи. Радикал гидроксила — небольшая по размеру незаряженная частица — способен проникать в толщу гидрофобного липидного слоя и вступать в химическое взаимодействие с полиненасыщенными жирными кислотами (которые принято обозначать как LH), входящими в состав биологических мембран и липопротеинов плазмы крови. При этом в липидном слое мембран образуются липидные радикалы:
НО- + LH -> Н20 + L
-
f
Липидный радикал (L-) вступает в реакцию с растворенным в среде молекулярным кислородом; при этом образуется новый свободный радикал — радикал липоперекиси (LOO):
L- + LH -> LOO .
Продолжение цепи. Этот радикал атакует одну из соседних молекул фосфолипида с образованием гидроперекиси липида LOOH и нового радикала L-*
LOO + LH LOOH + L-.
Чередование двух последних реакций представляет собой цепную реакцию перекисного окисления липидов (см. рис. 2.7, А).
Разветвление цепи. Существенное ускорение пероксидации липидов наблюдается в присутствии небольших количеств ионов двухвалентного железа. В этом случае происходит разветвление цепей в результате взаимодействия Fe2+ с гидроперекисями липидов:
Fe2+ + LOOH -> Fe3+ + НО- + LO-.
Образующиеся радикалы LO- инициируют новые цепи окисления липидов (рис. 2.7, Б):
LO + LH -> LOH + L-; L- + 02 -> LOO- -> и т.д.
Обрыв цепей. В биологических мембранах цепи могут состоять из десятка и более звеньев. Но в конце концов цепь обрывается в результате взаимодействия свободных радикалов с антиоксидантами (InH), ионами металлов переменной валентности (например, теми же Fe2+) или друг с другом:
LOO- + Fe2+ + Н+ -> LOOH;
LOO- + InH -> In-;
LOO- + LOO- -> молекулярные продукты.
Использование хемилюминесценции для изучения реакций, идущих с участием свободных радикалов. Последняя реакция интересна еще и тем, что она сопровождается свечением — хемилюминесценци- ей. Интенсивность хемилюминесценции очень мала, поэтому ее иногда называют «сверхслабым свечением». Интенсивность свечения пропорциональна квадрату концентрации свободных радикалов в мембранах, а скорость перекисного окисления прямо пропорциональна концентрации тех же радикалов. Поэтому интенсивность «сверхслабого» свечения однозначно отражает скорость липидной пероксидации в изучаемом биологическом материале, и измерение хемилюминесценции довольно часто используется при изучении перекисного окисления липидов в различных объектах.Измерение хемилюминесценции широко применяется также для изучения образования активных форм кислорода клетками крови и пери- тонеальными макрофагами. В присутствии специальных соединений — люминола и люцигенина — наблюдается хемилюминесценция изолированных лейкоцитов крови, макрофагов или разведенной цельной крови, если клетки-фагоциты продуцируют гипохлорит, и радикалы кислорода (супероксид + гидроксил-радикал). Интенсивность хемилюминесцентных ответов клеток увеличивается в несколько раз при появлении очагов некроза в организме, например после инфаркта миокарда, и, напротив, угнетается при тканевой гипоксии; поэтому измерение клеточной хемилюминесценции может быть использовано в ряде случаев с целью выявления заболевания, оценки тяжести состояния больного и эффективности назначенного лечения.
Биологические последствия пероксидации липидов. Увеличенное образование свободных радикалов в организме и связанное с этим усиление процессов пероксидации липидов (которое иногда называют оксидативным стрессом) сопровождается рядом нарушений в свойствах биологических мембран и функционировании клеток. Наиболее изучены три прямых следствия перекисного окисления липидов.
Во-первых, перекисное окисление липидов сопровождается окислением тиоловых(сульфгидрильных) групп мембранных белков (Рг). Это может происходить в результате неферментативной реакции SH-групп со свободными радикалами липидов; при этом образуются сульфгидриль- ные радикалы, которые затем взаимодействуют с образованием дисульфидов либо окисляются кислородом с образованием производных суль- фоновой кислоты:
Pr-SH + L- LH + Pr-S-;
PiyS- + Pr2-S- PiySS-Piv, Pr-S- + 02 -> Pr-S02* -> молекулярные производные.
Связанные с перекисным окислением липидов окисление белков и образование белковых агрегатов в хрусталике глаза заканчиваются его помутнением; этот процесс имеет большое занчение в развитии старческой и других видов катаракты у человека. Важную роль в патологии клетки играет также инактивация ион-транспортных ферментов, в активный центр которых входят тиоловые группы, в первую очередь Са2+—АТФазы. Инактивация этого фермента вызывает замедление «откачивания» ионов кальция из клетки и, наоборот, вход кальция в клетку (рис. 2.8, 7), увеличение внутриклеточной концентрации ионов кальция и повреждение клетки.
2(V (.V
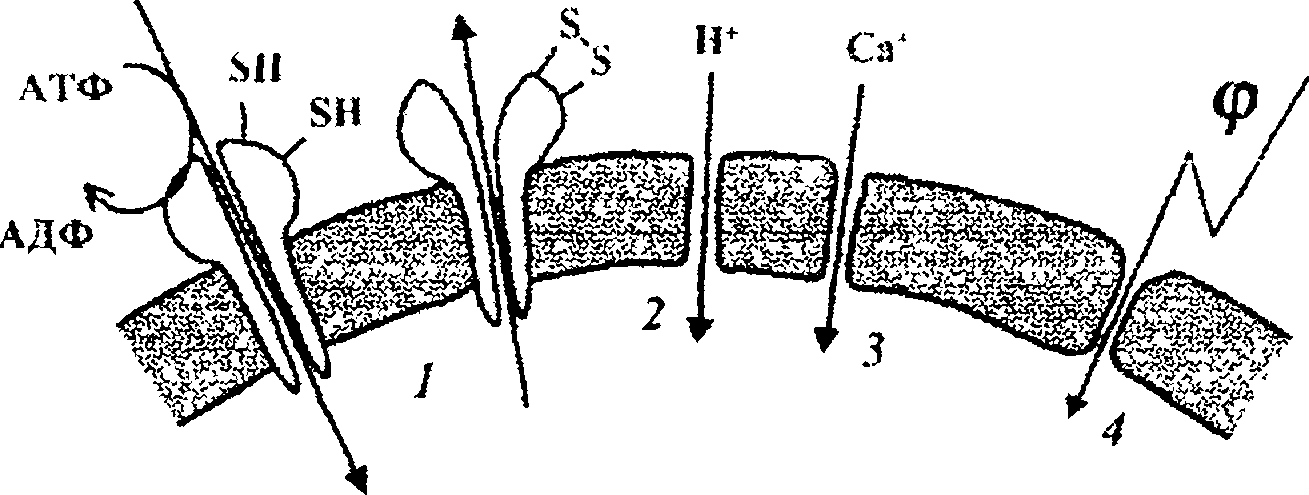
Рис. 2.8. Нарушение барьерных свойств мембран при перекисном окислении липидов.
Наконец, окисление тиоловых групп мембранных белков приводит к появлению дефектов в липидном слое мембран клеток и митохондрий. Под действием разности электрических потенциалов на мембранах через такие дефекты в клетки входят ионы натрия, а в митохондрий — ионы калия. В результате увеличивается осмотическое давление внутри клеток и митохондрий, что способствует еще большему повреждению мембран.
Во-вторых, результат перекисного окисления липидов связан с тем, что продукты пероксидации обладают способностью непосредственно увеличивать ионную проницаемость липидного бислоя. Показано, что продукты перекисного окисления липидов делают липидную фазу мембран проницаемой для ионов водорода (рис. 2.8, 2) и кальция (рис. 2.8, 3). Это приводит к тому, что в митохондриях окисление и фосфорилирование разобщаются, а клетка оказывается в условиях энергетического голода (т.е. недостатка АТФ). Одновременно в цитоплазму выходят ионы кальция, которые повреждают клеточные структуры.
Третьий (и быть может, самый важный) результат пероксидации — это уменьшение стабильности липидного слоя, что может вызвать электрический пробой мембраны собственным мембранным потенциалом, т.е. под действием разности электрических потенциалов, существующей на мембранах живой клетки. Электрический пробой приводит к полной потере мембраной ее барьерных функций (рис. 2.8, 4).
Клеточные системы антирадикальной защиты. В нормальных условиях процесс перекисного окисления липидов находится под строгим контролем ферментативных и неферментативных систем клетки, отчего скорость его невелика. Принято делить химические соединения и физические воздействия, влияющие на скорость перекисного окисления липидов, на прооксиданты (усиливают процессы перекисного окисления) и антиоксиданты (тормозят перекисное окисление липидов). К проокси- дантам в живой клетке относятся высокие концентрации кислорода (например, при длительной гипербарической оксигенации больного), ферментные системы, генерирующие супероксидные радикалы (например, ксантиноксидаза, ферменты плазматической мембраны фагоцитов и др.), ионы двухвалентного железа.
Хотя сам процесс перекисного окисления развивается в виде цепных реакций в липидной фазе мембран и липопротеинов, начальные (а возможно, и промежуточные) стадии этой сложной системы реакций протекают в водной фазе. Часть защитных систем клетки также локализуется в липидной, а часть — в водной фазах. В зависимости от этого можно говорить о водорастворимых и гидрофобных антиоксидантах.
Антиоксиданты водной фазы. Основные реакции в водной фазе, предшествующие цепному окислению, и роль антиоксидантов в ограничении скорости этих процессов можно представить в виде схемы:Супероксид- Каталаза,
дисмутаза пероксидазы
■О,
1Г>
jp рез+
Ловушки
Fe3+
м>
Ре2+
J
О,
с/г
Ко м п л е кс о н ы Цепные
реакции
Непосредственными предшественниками гидроксильного радикала, инициирующего цепное окисление липидов, служат ионы двухвалентного железа и перекись водорода (или образующийся из нее гипохлорит). По этой причине образование радикала гидроксила и пероксидация липидов тормозятся веществами, снижающими концентрацию одного из этих двух соединений. К ним относятся следующие вещества:
фермент супероксиддисмутаза — снижает концентрацию супероксидных радикалов и тем самым препятствует восстановлению ими ионов трехвалентного железа до двухвалентного. В клетке ионы же- - леза хранятся в трехвалентном состоянии в специальных депо, образованных субъединицами белка — ферритина;
ферменты каталаза и глутатионпероксидаза — удаляют перекись водорода. Эффективность работы глутатионпероксидазы зависит от концентрации свободного глутатиона, при снижении которой может возрастать концентрация цитотоксических гидроксильных радикалов;
регенерация восстановленного глутатиона (GSH) из окисленного (GSSG) осуществляется за счет НАДФН; этот процесс катализируется ферментом глутатионредуктазой. Недостаток глутатиона в клетках, например в эритроцитах, который может быть обусловлен действием токсичных веществ, например ионов тяжелых металлов или наследственным недостатком глутатионредуктазы, приводит к активации перекисного окисления; это, в частности, наблюдается при некоторых видах гемолитических анемий;
соединения, связывающие ионы железа (комплексоны). Следует, однако, добавить, что в водной фазе некоторые комплексы ионов железа вступают в реакции с супероксидным радикалом и перекисью водорода наряду со свободными ионами железа.
Антиоксиданты, тормозящие развитие цепных реакций в липидной фазе. Основные реакции в липидной фазе биологических мембран и липопротеинов крови, а также роль антиоксидантов в ограничении скорости этих процессов можно продемонстрировать с помощью схемы
.Ловушки радикалов
LH | 02 J LH 102 Цепь окислеиия
HO^jr^ L»-*-^ LOO L* ^— **
PC
О
CsJ
5
cs
LO*
^
LOH
Fe2
Комплексоны
I LH
loo-Ц
LOO
♦ tt
Фосфолипаза^
НООН
GSH-пероксидаза
Fe5+
к
о.
*
Цепные реакции «ведут» свободные радикалы липидов (L* и LOO), разветвление цепей происходит при взаимодействии продукта пероксидации — гидроперекиси липидов (LOOH) с ионами Fe2+. Все соединения, снижающие концентрацию перечисленных веществ, выполняют функцию антиоксидантов. К ним относятся:
ферменты фосфолипаза и глутатионпероксидаза, разрушающие гидроперекиси липидов, предотвращая разветвление цепей окисления липидов в мембранах. При этом действие фосфолипазы заключается в отщеплении от фосфолипидов окисленной жирной кислоты, содержащей гидроперекисную группу (LOOH), а действие глутатионпероксидазы сводится к восстановлению этой группы до спиртовой с одновременным окислением глутатиона (GSH) до дисульфида (GSSG):
LOOH + 2GSH LOH + GSSG + Н20;
ловушки радикалов, которые называют иногда «липидными анти- оксидантами». По своей химической природе липидные антиоксиданты — это производные фенола. К ним относится ос-токоферол (витамин Е), убихинон (кофермент Q), тироксин, эстрогены и синтетические соединения,например ионол;
он
CH-,
сн,
сн,
сн,
Ионол![]()
![]()
сн.
н,с
a-Tocopherol (витамин Е
)
— соединения, связывающие железо. Большинство из них, включая такие природные соединения, как дипептид карнозин, не просто связывают железо, но, главное, не дают ему возможности приникнуть в липидную фазу мембран, поскольку образующиеся комплексы в силу своей полярности не проникают в гидрофобную зону.
Для детоксикации двухвалентного железа в организме существует, по-видимому, целая система окисления и связывания ионов железа. В плазме крови эта система представлена ферментом церрулоплазмином (феррооксидазой), который окисляет Fe2+ до Fe3+ кислородом без образования свободных радикалов, и белком трансферрином, который связывает и переносит в кровяном русле ионы трехвалентного железа, а затем захватывается клетками. В клетках железо может восстанавливаться аскорбиновой кислотой и другими восстановителями, но затем окисляется и депонируется в окисленной форме внутри ферментного белкового комплекса ферритина.