


РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.2. ВВЕДЕНИЕ ЧУЖЕРОДНОЙ ДНК В КЛЕТКИ ДРОЖЖЕЙ И БАКТЕРИЙ
7.Пробирки вынуть изо льда, добавить по 250 мкл свежей LB среды (стерильно!) и инкубировать при 37 °С в течение 30 мин.
8.Высеять полученные клетки на соответствующие чашки с агаром. Для этого отобрать по 100 мкл клеточной суспензии и перенести ее на поверхность агара. Растереть стерильной стеклянной петлей досуха, закрыть чашки,
перевернуть и поместить в термостат (37 °С) на ночь.
9.Проанализировать полученные результаты. Сравнить количество выросших колоний на чашках с антибиотиком и без него.
10.Определите эффективность трансформации по формуле:
Эфф. трансф. = число колоний на чашке/кол-во ДНК на чашку (мкг).
Задание 1.2.2. Трансформация клеток E. сoli электропорацией
Порядок выполнения работы
1.Подписать две пластиковые пробирки: +ДНК и –ДНК.
2.В ламинарном боксе стерильно отобрать по 80 мкл суспензии электрокомпетентных клеток в воде в каждую пробирку. Поместить пробирки в ледяную баню.
3.В пробирку +ДНК внести 1 мкл раствора плазмидной ДНК в воде. В пробирку –ДНК внести такое же количество буфера без ДНК. Перемешать.
4.Перенести смесь клеток с ДНК в электропорационную кювету 0,1 мм.
5.Провести процедуру электропорации при пульсе 1800 кВ согласно инструкции к прибору.
6.Сразу же добавить к клеткам SOC среду и поместить их в термостат на 30 мин при 37 °С, лучше с перемешиванием.
7.Высеять полученные клетки на соответствующие чашки с агаром. Для этого отобрать по 100 мкл клеточной суспензии и перенести ее на поверхность агара. Растереть стерильной стеклянной петлей досуха, закрыть чашки,
перевернуть и поместить в термостат (37 °С) на ночь.
9.Проанализировать полученные результаты. Сравнить количество выросших колоний на чашках с антибиотиком и без него.
10.Определить эффективность трансформации и сравнить ее с полученной для химически компетентных клеток.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
35 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.2. ВВЕДЕНИЕ ЧУЖЕРОДНОЙ ДНК В КЛЕТКИ ДРОЖЖЕЙ И БАКТЕРИЙ
Задание 1.2.3. Трансформация клеток E. сoli XL1-Blue плазми-
дой pGLO
Цель работы – дать представление о процедуре генетической трансформации. Провести эксперимент по трансформации клеток E. сoli, продемонстрировать связи ДНК→РНК→БЕЛОК→СВОЙСТВО организма
В рамках данной работы бактериальные клетки E. coli (XL1-Blue) трансформируют плазмидой с геном, кодирующим зеленый флюоресцентный белок (GFP, green fluorescent protein), выделенным из биолюминесцирующей медузы Aequorea victoria. Благодаря этому белку медузы в природе флюоресцируют зеленым цветом. Трансформированные бактериальные клетки, экспрессирующие ген GFP, продуцируют зеленый флюоресцентный белок и флюоресцируют зеленым цветом при облучении ультрафиолетовой лампой.
Материалы и оборудование:
1.Компетентные клетки E. coli (XL1-Blue).
2.Раствор плазмидной ДНК (pGLO).
3.Трансформационный буфер.
4.Агар на LB среде.
5.Ампициллин, р-р 200 мг/мл.
6.Арабиноза, 20 %-й раствор.
7.Чашки Петри.
8.Пипетки, стерильные типсы.
9.Пластиковые пробирки Eppendorf.
10.Стеклянные петли.
11.Центрифуга.
12.Контейнер со льдом, водяная баня (42 оС), термостат (37 оС).
13.Микробиологический бокс.
14.UV-лампа.
Порядок выполнения работы
1.Подписать две пластиковые пробирки: +ДНК и –ДНК.
2.В боксе стерильно отобрать по 100 мкл суспензии компетентных клеток в трансформационном буфере в каждую пробирку. Поместить пробирки
вледяную баню.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
36 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.2. ВВЕДЕНИЕ ЧУЖЕРОДНОЙ ДНК В КЛЕТКИ ДРОЖЖЕЙ И БАКТЕРИЙ
3.В пробирку +ДНК внести 1 мкл раствора плазмидной ДНК. В проби р- ку –ДНК внести такое же количество буфера без ДНК. Выдержать обе смеси на льду не менее 20 мин.
4.Чашки Петри подписать на дне следующим образом LB-ДНК, LB/аmp-
ДНК, LB/аmp+ДНК, LB/amp/ara-ДНК, LB/amp/ara+ДНК.
5.Расплавить агар в микроволновой печи, перенести его в бокс.
6.На дно соответствующих чашек поместить растворы ампициллина (0,25 мл, конечная конц. 200 мкг/мл и арабинозы, (0,25 мл, конечная конц. 0,2%). Залить в чашки агар (по 25 мл), осторожно перемешать и оставить полимеризоваться в боксе под UV-облучением.
7.Провести процедуру теплового шока. Для этого обе пробирки поместить в водяную баню (42оС) на 25 сек (строго!), после чего быстро перенести их опять на лед. Выдержать 2-3 минуты.
8.Пробирки вынуть изо льда, добавить по 500 мкл свежей LB среды (стерильно!) и инкубировать при 37оС в течение 30 мин.
9.Высеять полученные клетки на соответствующие чашки с агаром. Для этого отобрать по 100 мкл клеточной суспензии и перенести ее на поверхность агара. Растереть стерильной стеклянной петлей досуха, закрыть чашки, перевернуть и поместить в термостат (37оС) на ночь.
Анализ полученных результатов:
1. Пронаблюдайте полученные результаты посева на чашках Петри при нормальном освещении и при облучении чашек ультрафиолетовой лампой. Внесите свои наблюдения в таблицу 1.1, отвечая на следующие вопросы:
•как много колоний выросло на чашке (сравнительно)?
•какого они цвета?
•что наблюдали при облучении чашек ультрафиолетом?
Таблица 1.1
+DNA, LB/amp
+DNA, LB/amp/ara
-DNA, LB/amp
-DNA, LB
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
37 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.2. ВВЕДЕНИЕ ЧУЖЕРОДНОЙ ДНК В КЛЕТКИ ДРОЖЖЕЙ И БАКТЕРИЙ
2.Опишите, какие новые свойства приобрели трансформированные бактерии. Благодаря чему?
3.Определите эффективность трансформации по формуле:
Эфф. трансф. = число колоний на чашке / кол-во ДНК на чашку (мкг)
4.Определите число колоний, выросшее на чашках (+DNA, LB/amp) и (+DNA, LB/amp/ara).
5.Количество ДНК определите исходя из концентрации раствора ДНК и его количества, взятого для трансформации. Запишите проведенные расчеты
иполученные результаты.
 Контрольные вопросы
Контрольные вопросы
1.Что такое генетическая трансформация?
2.Что такое компетентные клетки? Как вы себе представляете процесс проникновения плазмидной ДНК внутрь клеток в момент температурного шока?
3.Почему для проведения генетических модификаций чаще всего используют клетки E.coli? Какие еще организмы используются в биотехнологии?
4.Какова эффективность проведенной вами трансформации? От чего она зависит? Почему клетки на чашке +DNA, LB/amp выросли, но не флюоресцируют?
5.Как вы думаете, о чем бы свидетельствовала флюоресценция клеток, выросших на чашке +DNA, LB/amp?
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
38 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА1.3. ЭКСПРЕССИЯГЕНОВ, КЛОНИРОВАННЫХВПРОКАРИОТИЧЕСКИХ СИСТЕМАХ. КУЛЬТИВИРОВАНИЕРЕКОМБИНАНТНЫХКЛЕТОКE.coli
 Цель лабораторной работы
Цель лабораторной работы
•культивирование рекомбинантных клеток E.coli BL21(DE3), с контролируемой экспрессией двух различных белков: рекомбинантного белка апообелина (плазмида рЕТ-OL8) и химерного белка proZZ-Obe (плазмида pTZZO2)
 Краткие теоретические сведения
Краткие теоретические сведения
Основная цель экспериментов по клонированию генов, которые предполагается использовать в биотехнологии, – подбор условий для эффективной экспрессии в нужном организме-хозяине. К сожалению, сам фактор встраивания того или иного гена в клонирующий вектор еще не означает, что этот ген будет эффективно экспрессирован. Среди молекулярнобиологических свойств систем экспрессии наиболее важны следующие:
•тип промотора и терминатора транскрипции;
•прочность связывания мРНК с рибосомой;
•число копий клонированного гена и его локализация (в плазмиде или в хромосоме хозяйской клетки);
•конечная локализация синтезируемого продукта;
•эффективность трансляции в организме хозяина;
•стабильность продукта в хозяйской клетке.
Универсальной стратегии оптимизации экспрессии генов не существует. Большинство таких генов, а также белки, которые они кодируют, имеют уникальные молекулярные свойства, и оптимальные системы экспрессии для каждого из них приходится подбирать каждый раз заново. К настоящему времени разработан целый ряд экспрессирующих систем, позволяющих экспрессировать множество различных белков. Одной из наиболее продуктивных систем является так называемая pET система на основе транскрипционных и трансляционных регуляторных элементов фага T7. Она включает в себя широкий спектр экспрессирующихся плазмид (векторов) и хозяйских штаммов E.coli, обладающих различными свойствами. Структурный ген в этой системе находится под контролем промотора РНК-полимеразы фага Т7. Это
|
Современные аппаратура и методы исследования биологических систем. Большой практикум.Учебебное пособие |
39 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.3. ЭКСПРЕССИЯ ГЕНОВ, КЛОНИРОВАННЫХ В ПРОКАРИОТИЧЕСКИХ СИСТЕМАХ
один из самых сильных промоторов среди изученных на сегодняшний день. Этот промотор не узнается геномной РНК-полимеразой E.coli и структурный ген под этим промотором не экспрессируется. Такая система особенно эффективна в случае экспрессии белков, токсичных для организма-хозяина или в случаях, когда экспрессия чужеродного белка приводит к метаболической перегрузке организма-хозяина.
Рис. 1.11. Структура экспрессирующей части pET-векторов
На рис. 1.11 схематически показана структура экспрессирующей части pETвекторов.
Для транскрипции с промотора фага Т7 требуется соответствующая РНК-полимераза, ген которой может быть встроен в геномную ДНК под ко н- тролем какого либо промотора. Фирмой Novagen разработано 9 таких штаммов E.coli , обладающих геном РНК полимеразы фага Т7 в геномной ДНК под контролем промотора lacUV5, представляющего собой усиленный вариант lac-промотора дикого типа и эффективно работающих как хозяйские организмы для экспрессии pET-векторов. На рис. 1.12 показан контролирующий комплекс элементов в системе рЕТ.
В предлагаемой работе нами используется штамм E.coli BL21(DE3). Помимо жесткой регуляции экспрессии этот штамм лишен ряда протеолитиче-
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
40 |
РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.3. ЭКСПРЕССИЯ ГЕНОВ, КЛОНИРОВАННЫХ В ПРОКАРИОТИЧЕСКИХ СИСТЕМАХ
ских ферментов, что способствует уменьшению протеолитического расщепления рекомбинантных белков в клетке-хозяине.
Рис. 1.12. Контролирующий комплекс элементов в системе рЕТ
 Материалы и оборудование
Материалы и оборудование
1.Трансформированные клетки.
2.Стерильная LB среда.
3.Стерильные колбы объемом 100 мл – 2 шт; 1 л – 2шт
4.Ампициллин (р-р, 200 мг/мл).
5.ИПТГ (изопропил –β-D-тиогалактопиранозид).
6.Микробиологический бокс.
7.Термостатированный шейкер.
8.Фотоэлектроколориметр (ФЭК) и кюветы к нему.
9.Центрифуга с охлаждением и центрифужные стаканы к ней.
10.Весы для уравновешивания центрифужных стаканов.
11.Набор автоматических пипеток и наконечников к ним.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
41 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.3. ЭКСПРЕССИЯ ГЕНОВ, КЛОНИРОВАННЫХ В ПРОКАРИОТИЧЕСКИХ СИСТЕМАХ
 Задание на выполнение лабораторной работы
Задание на выполнение лабораторной работы
Задание 1.3.1 Культивирование рекомбинантных клетокE.coli BL21(DE3), с контролируемой экспрессией двух различных белков
Порядок выполнения работы
Занятие № 1
1.Подписать колбы для культивирования «Obe» и «proZZ-Obe».
2.В стерильных условиях внести в 100мл колбы по 20 мл стерильной LBсреды, раствор ампициллина до конечной концентрации 200 мкг/мл и 2-3 среднего размера колонии соответствующих бактерий. Инкубировать при 37 оС при активном перемешивании до плотности Д590= 0,5-0,6. Оптическую
плотность измерять через каждые 45-60 мин с помощью ФЭКа против исходной среды. Пробу отбирать стерильными наконечниками.
3. Полученную культуру поместить в холодильник до следующего утра.
Занятие № 2
1.Отцентрифугировать клетки с помощью центрифуги К-23 ( 3–4 тыс. об/мин., 5 оС, 15 мин).
2.В колбы объемом 1л с 200 мл стерильной LB-среды внести раствор ампицилина до конечной концентрации 200 мкг/мл. Стерильно перенести клетки в эти колбы, суспендируя их свежей стерильной LB-средой.
3.Инкубировать при 37 оС при активном перемешивании. Следить за
ростом клеток, измеряя оптическую плотность ОД590 против исходной среды через каждый час.
4.При плотности ОД590 = 0,6-0,8 отобрать по 1 мл культуры и отцентрифугировать на микроцентрифуге. Хранить образец клеток до индукции при
минус 20 оС. Провести индукцию транскрипции рекомбинантного белка. Для этого в колбы добавить сухую навеску ИПТГ (до концентрации 100 мг/л).
5. Продолжить культивирование еще в течение 3 ч, следить за ростом, как описано в п.3. Отобрать аликвоту клеточной суспензии, равную количеству клеток до индукции, отцентрифугировать и хранить образец, как описано в п. 4.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
42 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.3. ЭКСПРЕССИЯ ГЕНОВ, КЛОНИРОВАННЫХ В ПРОКАРИОТИЧЕСКИХ СИСТЕМАХ
6.Построить кривую роста оптической плотности культуры (при Д590) во времени.
7.Бактерии отделить центрифугированием, как описано в п. 1.
8.Промыть клетки ресуспендируя физраствором (0,9 %-й NaCl) и вновь отцентрифугировать. Взвесить полученную клеточную пасту.
Хранить клетки при минус 20 оС до следующего занятия.
 Контрольные вопросы
Контрольные вопросы
1.Для чего нужна регуляция экспрессии рекомбинантных белков?
2.Назовите регуляторные участки, которые должны присутствовать в экспрессируемой плазмиде.
3.Ген экспрессируемого в Е.coli белка находится под промотором РНК полимеразы фага Т7. Какие гены должны присутствовать в клетке-хозяине, чтобы экспрессия была эффективной?
4.Каковы свойства клеток Е.coli BL21(DE3), которые вы использовали для экспрессии рекомбинантных белков?
5.Опишите регуляторные механизмы экспрессии в системе рЕТ, пользуясь рис. 1.11. Нарисуйте схематически плазмиды для экспрессии апообелина и химерного белка.
6.Что такое ИПТГ? Что происходит в трансформированных клетках после его добавления в питательную среду?
7.В каких условиях производится культивирование трансформирован-
ных клеток Е.coli BL21(DE3)?
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
43 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА1.4. ВЫДЕЛЕНИЕРЕКОМБИНАНТНЫХБЕЛКОВАПООБЕЛИНАИ
PROZZ-OBE
 Цель лабораторной работы
Цель лабораторной работы
•выделение рекомбинантного апообелина и химерного белка proZZ-Obe из биомассы полученных трансформированных клеток
E. coli
 Краткие теоретические сведения
Краткие теоретические сведения
При высокой экспрессии рекомбинантных белков, как правило, клеткихозяева «пакуют» их в виде не растворимых частиц, так называемых телецвключений. Молекулы белка в тельцах-включениях не обладают определенной третичной структурой и специфической активностью. При этом тельца включения содержат экспрессированный белок практически в чистом виде. Находясь в не растворенном виде, он практически не подвергается протеолизу. С одной стороны, это существенно упрощает процедуру очистки целевого белка, но с другой – возможно возникновение проблемы его посттрансляционного фолдинга (т.е. формирования правильной, функционально активной третичной структуры). Особенно остро эта проблема может возникнуть, например, когда целевой белок должен образовывать правильные S-S- мосты. Существует ряд приемов, позволяющих решить проблему фолдинга. Например, транспорт рекомбинантного белка в периплазматическое пространство или в межклеточную среду, либо использование специфических ферментов, так называемый фолдаз.
Из телец-включений рекомбинантные белки переводятся в раствор с помощью высоких концентраций хаоторопных агентов – мочевины или гуа- нидин-хлорида. В таких растворах белки также не обладают активной структурой, однако могут быть дополнительно очищены, например хроматографическими методами.
В случае кальций-зависимых фотопротеинов, апообелина в частности, проблемы фолдинга не возникает. Апообелин – односубъединичный, сранительно небольшой белок, не обладающий S-S-мостиками. Формирование третичной структуры активного обелина происходит на стадии его активации субстратом – целентеразином. Образование телец-включений апообелина существенно упрощает выделение и очистку этого белка.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум.Учебебное пособие |
44 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.4. ВЫДЕЛЕНИЕ РЕКОМБИНАНТНЫХ БЕЛКОВ АПООБЕЛИНА И PROZZ-OBE
 Материалы и оборудование
Материалы и оборудование
1.Биомасса трансформированных клеток E. coli.
2.Буферные растворы для промывки телец-включений: а) 20 мМ раствор Трис-HCl, рН 7,0 – 100 мл;
б) 0,9 %-й раствор NaCl в а) –20 мл;
в) 0,1 %-й раствор Triton X-100 в а) – 20 мл;
г) 5 мМ раствор CaCl2 в а) –20 мл;
д) 6 М мочевина, 20 мМ Трис-HCl, рН 7,0, 5 мМ CaCl2 – 20 мл.
3.Все перечисленные растворы готовятся студентами из соответствующих концентрированных растворов.
4.Ультразвуковой дезинтегратор.
5.Микроцентрифуга для пробирок «Eppendorf» 5417R (США) c ротором для микропробирок 1,5-2 мл.
6.Весы для уравновешивания центрифужных стаканов.
7.Набор автоматических пипеток и наконечники к ним.
 Задание на выполнение лабораторной работы
Задание на выполнение лабораторной работы
Задание 1.4.1. Выделение рекомбинантногоапообелина ихимерного белкаproZZ-Obe избиомассы полученных трансформированных клеток E. coli
Порядок выполнения работы
1.Ресуспендировать клетки в пятикратном объеме 20 мМ р-ре ТрисHCl, рН 7,0 (вес/объем) и разрушить озвучиванием 5х20 с при охлаждении льдом.
2.Полученную смесь центрифугировать (4-5 тыс. об/мин., 20 мин).
3.Супернатант отбросить, а осадок ресуспендировать в таком же объеме раствора б) и вновь отцентрифугировать.
4.Повторить процедуру п. 3, используя для промывки осадка последовательно растворы в) и г).
5.Полученный осадок ресуспендировать в том же объеме р-ра д) и оставить при перемешивании на ночь при 4 оС.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
45 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.4. ВЫДЕЛЕНИЕ РЕКОМБИНАНТНЫХ БЕЛКОВ АПООБЕЛИНА И PROZZ-OBE
 Контрольные вопросы
Контрольные вопросы
1.Какие методы используют для разрушения бактериальных клеток?
2.Что такое “тельца включения”? Какие преимущества и недостатки они дают при выделении рекомбинантных белков?
3.Для чего промывают тельца-включения каждым из растворов а)-г)?
4.Как переводят белки из телец-включений в раствор?
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
46 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА1.5. ОПРЕДЕЛЕНИЕНУКЛЕОТИДНОЙПОСЛЕДОВАТЕЛЬНОСТИДНКНА СЕКВЕНАТОРЕALFexpress II DNA
 Цель лабораторной работы
Цель лабораторной работы
•изучение порядка экспериментального определения нуклеотидной последовательности ДНК
 Краткие теоретические сведения
Краткие теоретические сведения
Знание последовательности ДНК необходимо для понимания молекулярных основ жизни. Порядок азотистых оснований (G, A, T или C) определяет структуру ДНК и ее функции, весь набор молекулярных программ развития, старения и смерти организма. Сравнивая последовательности ДНК из разных организмов, возможно реконструировать эволюцию жизни на Земле. Систематика живых организмов и филогенетические взаимосвязи в настоящее время пересматриваются с учетом данных по нуклеотидным последовательностям.
Методы определения порядка нуклеотидных оснований в молекуле ДНК в отечественной литературе принято называть методами секвениро-
вания. Используемые методы основаны либо на химической деградации нуклеиновой кислоты, либо, что более широко распространено, на энзиматическом синтезе секвенируемого района. За создание обоих методов авторам была присуждена Нобелевская премия.
Автоматизации секвенирования ДНК позволила перейти к расшифровке генетической информации целых организмов. В последние годы наблюдается экспоненциальный рост числа известных нуклеотидных последовательностей. Первый вирус был секвенирован в 1980-х гг., первая бактерия – в 1995, первый многоклеточный организм (Caenorhabditis elegans) – в 1998 г., нуклеотидная последовательность 95% генома человека была впервые представлена в 2000 г. К 2004 г. было расшифровано 3 генома млекопитающих: человека, мыши и крысы, не считая полного прочтения многих геномов других организмов. Большая часть информации по секвенированию хранится в публичных базах данных и доступно каждому через Интернет.
Секвенирование ДНК по методу Максама и Гилберта: метод химиче-
ской деградации. В 1976 г. А. Максамом и У. Гилбертом был разработан м е- тод секвенирования, основанный на сайт-специфической химической деградации фрагмента ДНК, радиоактивно меченного с одного конца. Препарат
|
Современные аппаратура и методы исследования биологических систем. Большой практикум.Учебебное пособие |
47 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
меченой ДНК разделяли на четыре аликвоты и каждую обрабатывали реагентом, модифицирующим одно или два из четырех оснований. А. Максам и У. Гилберт предложили модифицировать пуриновые основания диметилсульфатом. При этом происходит метилирование адениновых (А) остатков по азоту в положении 3, а гуаниновых (G) – по азоту в положении 7. Обработка образца ДНК соляной кислотой при 0 °С приводит к выщеплению метиладенина. Последующая инкубация при температуре 90 °С в щелочной среде вызывает разрыв сахарно-фосфатной цепи ДНК в местах выщепления оснований. Обработка пиперидином приводит к гидролизу образца по остаткам метилгуанина. Пиримидиновые основания (C и Т) модифицируют гидразином. Если реакцию вести в бессолевой среде, то модифицируются как цитозин (С), так и тимидин (Т); если обработку вести в присутствии 2 М NaCl, то модифицируется лишь цитозин. Расщепление цепи ДНК на фрагменты и в этом случае осуществляется пиперидином. Условия реакций авторы подбирали таким образом, чтобы в итоге получить полный набор частично расщепленных субфрагментов разной длины. Электрофорез в полиакриламидном геле позволяет восстановить полную структуру исследуемого фрагмента.
Секвенирование по методу Максама-Гилберта не является рутинной методикой, поскольку используются токсичные реагенты, кроме того, метод довольно трудоемок. Сегодня химическое секвенирования применяется в основном для синтетических олигонуклеотидов, которые не могут быть проанализированы методом ферментативого синтеза.
Ферментативное секвенирование ДНК по методу Сэнгера: метод тер-
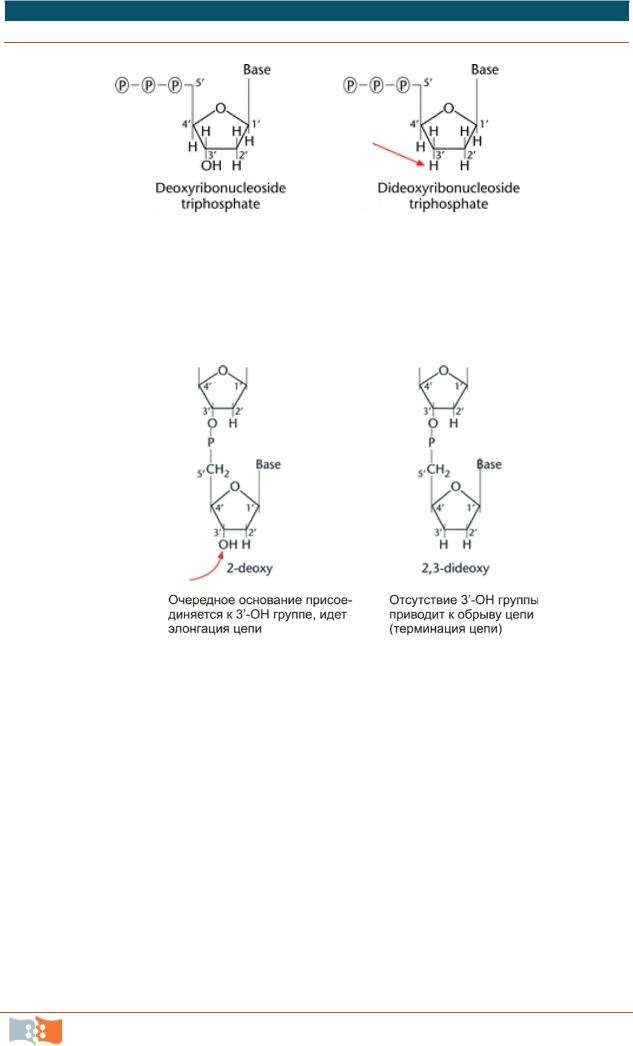
минаторов. В 1977 г. Ф. Сэнгер и его коллеги предложили способ ферментативного секвенирования, получивший название метода терминаторов. В основе метода лежало ферментативное копирование ДНК с помощью фрагмента Кленова ДНК полимеразы I из E. сoli. В качестве праймеров использовали синтетические олигонуклеотиды. В разделенном на четыре части образце проводили специфическую терминацию синтеза по каждому из оснований добавлением в реакционную смесь помимо четырех типов dNTP одного из аналогов трифосфатов – 2',3'-дидезоксинуклеозидтрифосфат (ddATP, ddTTP, ddCTP или ddGTP) (рис. 1.13–1.14). Дидезоксинуклеозидтрифосфат встраивался в синтезируемую цепь ДНК и блокировал ее дальнейший рост из-за отсутствия 3’-гидроксильной группы (3'-ОН).
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
48 |
РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
Рис. 1.13. Дезоксинуклеозидтрифосфат и его аналог дидезоксинуклеозидтрифосфат. Отсутствующая 3’-OH группа, необходимая для элонгации цепи ДНК, указана стрелкой
Рис. 1.14. Блокирование полимеризации ДНК при включении в растущую цепь дидезоксинуклеозидтрифосфата. Стрелкой указано место присоединения очередного трифосфата к растущей цепи ДНК
Отношение концентраций dNTP/ddNTP авторы подбирали экспериментально так, чтобы в итоге получить набор копий ДНК различной длины. Радиоактивную метку в синтезируемые цепи вводили использованием в реакции меченного по альфа положению фосфата одного из dNTP.
После этого полученные продукты четырех реакций копирования разгонялись в полиакриламидном геле на соседних дорожках, с геля получали радиоавтограф и по расположению полос определяли последовательность нуклеотидов (рис. 1.15).
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
49 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
С различными модификациями данный метод широко используется и в настоящее время.
Автоматическое секвенирование ДНК. В основе автоматического сек-
венирования ДНК преимущественно лежит уже упоминавшийся выше метод ферментативного секвенирования с использованием терминирующих ddNTP (другие альтернативные подходы: пиросеквенирование и секвенирование с помощью чип-гибридизации, не так широко распространены). Автоматизация стала возможной с введением в практику флюоресцентных меток.
Как и классический вариант Сэнгера, автоматическое секвенирование включает две стадии: проведение терминирующих реакций и разделение продуктов этих реакций с помощью электрофореза. Как правило, автоматизирована лишь вторая стадия, т. е. разделение меченных фрагментов ДНК в полиакриамидном геле, получение спектра эмиссии флуорофоров и анализ данных. Таким образом, автоматическое секвенирование методически отличается от ручного секвенирования только типом используемой метки.
Флуоресцентную метку включают либо в праймер, либо в терминатор транскрипции (ddNTP) согласно следующим схемам: 1) меченый праймер (четыре разных красителя) и немеченые терминаторы; 2) меченый праймер (один краситель) и немеченые терминаторы; 3) меченые терминаторы (каждый тип терминатора своим красителем) и немеченый праймер. Использование меченных праймеров предполагает проведение четырех независимых реакций (отдельно с каждым из терминаторов) для каждого секвенируемого образца. Использование четырех разных меток для терминаторов позволяет совместить все четыре реакции в одной пробирке. Если используется единственный краситель, то проводится четыре независимых реакции (отдельно с каждым из терминаторов) и разделение продуктов сиквенсовой реакции в геле выполняют на четырех разных дорожках (рис. 1.16). Использование четырех разных красителей позволяет разгонять продукты реакций на одной дорожке.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
50 |
РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
Рис. 1.15. Схема реакции ферментативного секвенирования с применением терминаторов
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
51 |
РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
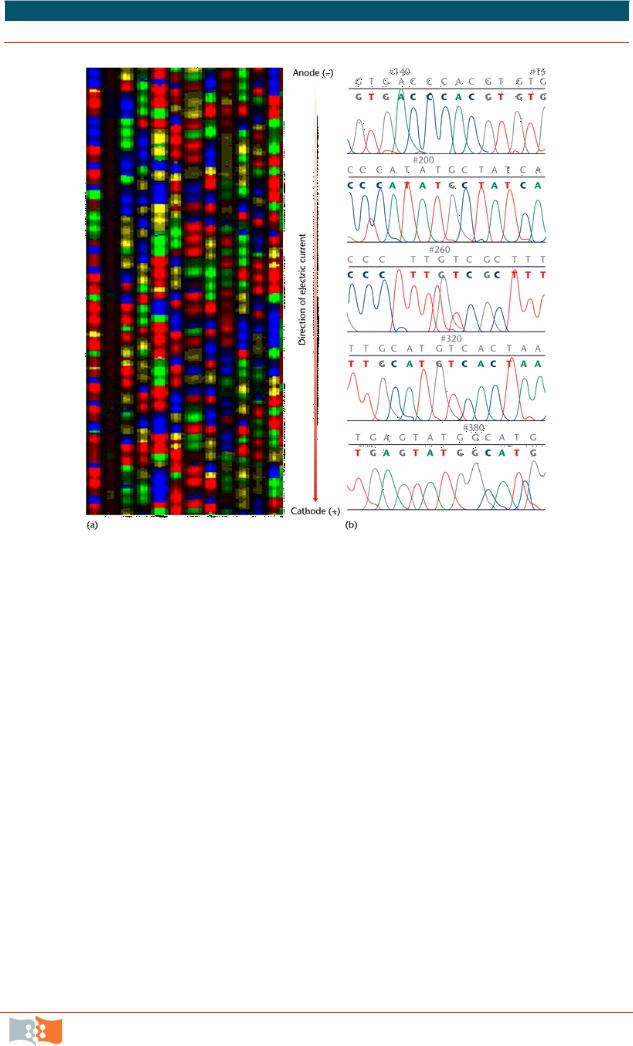
Рис. 1.16. Снимаемые данные с автоматического секвенатора Перкин-Элмер ABI PRISM 377. Необработанные секвенсовые данные, представленные в виде геля (a), и обработанные данные в виде фрагмента хроматограммы. Каждая четырехцветная вертикальная дорожка на левом рисунке соотвествует одной секвенсовой реакции
Флуоресцентные красители.
Флуоресценция – это излучение, сопровождающее переход электрона из возбужденного состояния в основное без изменения мультиплетности (т. е. переход синглет-синглет, либо триплет-триплет). Типичное время излучения для флуоресценции 10-8 с. Флуоресценции должен предшествовать обратный процесс – переход электрона в возбужденное состояние в результате поглощения (абсорбции) кванта света. Для флуоресценции доказана справедливость следующих правил: спектр испускания (эмиссии) не зависит от длины волны возбуждения; испускание сдвинуто относительно поглощения в сторону больших длин волн из-за энергетических потерь (сдвиг Стокса);
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
52 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
спектр испускания представляет собой зеркальное отражение спектра поглощения.
Спектр абсорбции, равно как и спектр эмиссии, зависит от химической структуры флуорофора, а также от условий, в которые молекула флуорофора помещена (рН, температура, среда и т. д.). В автоматическом секвенировании используют флуорофоры, абсорбция и излучение у которых происходит в диапазоне длин волн 450 –650 нм (видимая область спектра; секвенаторы ABI и Pharmacia) и 650–825 нм (ближняя инфракрасная область спектра; секвенаторы фирмы LI-COR).
К настоящему времени синтезировано большое число разнообразных флуоресцентных красителей и постоянно продолжается работа над новыми, с улучшенными характеристиками. Помимо высокого квантового выхода флуоресценции красители должны удовлетворять двум требованиям, перечисленным ниже. Присоединение молекулы флуорофора способно изменять подвижность меченого фрагмента ДНК в геле. Если при этом используются одновременно несколько красителей, то необходимо, чтобы влияние каждого из них на подвижность было либо минимальным, либо одинаковым у всех. Выравнивание электрофоретической подвижности (когда это нужно) проводят с помощью линкерных молекул, встраиваемых между красителем и, например, праймером. Еще одним важным моментом является условие минимального перекрывания спектров эмиссии. К сожалению, полностью избежать перекрывания спектров до сих пор не удалось.
Полимеразы, используемые при секвенировании ДНК. В оригиналь-
ной работе Ф. Сэнгера для проведения сиквенсовых реакций быд взят кленовский фрагмент ДНК-полимеразы I из E. сoli. В настоящее время для секвенирования используют рекомбинантные ДНК-полимеразы, отвечающие следующим требованиям: отсутствие 3'- и 5'-экзонуклеазной активности, отсутствие дискриминации по включению в растущую цепь как обычных, так и модифицированных (меченных) ddNTP. Выбор конкретной полимеразы зависит от условий проведения сиквенсовой реакции. Существует два разных подхода.
1. Копирование осуществляется при 37 °С высокопроцессивными термолабильными полимеразами, наиболее часто используется T7 ДНКполимераза и ее мутантные формы. Реакция требует одноцепочечной ДНК в качестве матрицы.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
53 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
2. Циклосеквенирование – реализуется циклический процесс полимеразной цепной реакции (односторонняя ПЦР), который включает денатурацию, отжиг и элонгацию, и предполагает использование термостабильных полимераз (в основном Tag-полимераза и ее производные под разными коммерческими названиями). Циклосеквенирование требует меньше матричной ДНК, в качестве которой может быть использована любая одноили двуцепочечная ДНК, а также резко уменьшает влияние потенциальных вторичных структур в ДНК, поскольку реакция идет при высоких температурах.
Секвенирующий гель и электрофорез. Полученные в реакции секве-
нирования радиоактивно/флуоресцентно меченные одноцепочечные фрагменты ДНК разделяют с помощью электрофореза в полиакриламидном геле. Гели, используемые в секвенировании, должны уметь разделять фрагменты, отличающиеся друг от друга на один нуклеотид в широком диапазоне длин. Разделение должно проходить в денатурирующих условиях, препятствующих ренатурации и возникновению вторичных структур у разделяемых фрагментов. В общем случае этим требованиям удовлетворяют 5-8 %-е полиакриламидные гели, содержащие 7 М мочевину или формальдегид. Обычно электрофорез проводится в трис-боратном буфере (89 мМ Трис-HCl, 8.9 мM борной кислоты, 2 мM ЭДТА, рН 8,0-8,5). Для эффективного разделения напряжение должно составлять 30-50 В на 1 см длины геля. Важным условием при проведении электрофореза является однородность температуры по всей поверхности геля. Неравномерный нагрев геля и стекол во время электрофореза способен привести к разнице в скоростях движения фрагментов ДНК по ширине геля и, как следствие, к искажению результатов. Для борьбы с этим явлением используют тонкие (обычно 0,4–0,1 мм) гели и принудительный подогрев стекол. В автоматическом секвенировании помимо электрофореза в пластинах полиакриламида чрезвычайно популярен капиллярный электрофорез в линейном полиакриламиде. Капилляры представляют собой стеклянную трубку длинной 30–100 см, закатанную в полимерный пластификатор. Небольшой диаметр капилляра (50–100 мкм) позволяет проводить разделение значительно быстрее, чем в обычных гелях. Кроме того, капиллярные секвенаторы позволяют обеспечивать гораздо более высокую чувствительность за счет отсутствия горизонтальной диффузии.
Таким образом, современные автоматические секвенаторы можно разделить по типу проводимого электрофореза на капиллярные и те, в которых разделение происходит в гелевых пластинах. Последние могут также
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
54 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
различаться по количеству детектируемых красителей: один, два или четыре. Капиллярные секвенаторы выпускаются только в варианте, использующем детекцию четырех флуоресцентных красок.
Секвенирование геномов. Типичная длина фрагментов ДНК, читаемых на автоматических секвенаторах, зависит от различных параметров и составляет от 500 до 1000 нуклеотидных оснований. Поскольку длина молекул ДНК в живых организмах несопоставимо больше, для полного прочтения генетической информации применяются две стратегии: направленное секвениро-
вание и рэндом-секвенирование, так называемый шотган-метод (shotgun).
В шотган-секвенировании большие фрагменты ДНК (типично больше 20 000 пар оснований) разбиваются на мелкие, которые вставляются в вектор. Предполагается, что сумма информации во всех этих клонах эквивалентна содержащейся в исходном ДНК-фрагменте. Большое количество маленьких клонов отбирается случайным образом, ДНК-матрица из этих клонов очищается и секвенируется. Далее, исходная последовательность большой исходной ДНК реконструируется компьютерной сборкой последовательностей из малых фрагментов. Эта стратегия интенсивно используется для прочтения больших геномов. Однако рэндом-стратегии обычно недостаточно для определения полного секвенса, поскольку какая-то маленькая часть фрагментов также случайно остается непрочтенной. И тогда используют направленное секвенирование для завершения проекта.
Направленное секвенирование, или прогулка по хромосоме, применяют для заполнения образующихся после рэндом-секвенировании пробелов или как оптимальный подход для секвенирования меньших ДНКфрагментов. Эта стратегия использует различные ДНК-праймеры, которые отжигаются на секвенируемой ДНК в единственном сайте и служат стартовой точкой для элонгации цепи/началом читаемого фрагмента. Для дизайна праймеров, c которых читаются неизвестные фрагменты, нужно иметь некоторую секвенсовую информацию. При этом первый фрагмент ДНК считается с известной уже последовательности, конец прочитанного фрагмента используется для дизайна нового праймера, с которого читается очередной неизвестный фрагмент, и цикл снова повторяется. Этот процесс минимизирует избыточность, но требует синтеза нового праймера для каждого раунда секвенирования.
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
55 |

РАЗДЕЛ 1. ГЕНЕТИЧЕСКАЯ ИНЖЕНЕРИЯ НИЗШИХ ОРГАНИЗМОВ
РАБОТА 1.5. ОПРЕДЕЛЕНИЕ НУКЛЕОТИДНОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК НА СЕКВЕНАТОРЕALFexpress II DNA
Необходимость дизайна и синтеза все новых праймеров, связанная с материальными и временными затратами, ограничивает рутинное применение направленного секвенирования.
 Материалы и оборудование
Материалы и оборудование
1.Секвенатор ALFexpress DNA Analysis System.
2.Раствор для приготовления геля.
3.Раствор bind-silane для обработки стекол кассеты геля.
4.Заливочный столик.
5.Буфер для электрофореза ТБЕ.
6.Деионизованная вода.
7.Кит для постановки сиквенсовой реакции.
8.Образцы плазмидной ДНК (матрицы).
9.Промывалка.
10.Фильтровальная бумага.
11.Спирт.
12.10 % -я уксусная кислота.
13.Пипетки 20 и 200 мкл и наконечники к ним.
14.Вытянутые наконечники для нанесения образцов на гель.
15.Стеклянные цилиндры 25 и 100 мл.
16.Пробирка 15 мл.
17.Вата.
18.Щприц с иголкой для промывания кармашков геля.
19.Деревянный молоточек.
20.Детергент (Fairy).
21.Губка для мытья стекол.
 Характеристики оборудования
Характеристики оборудования
 СистемаавтоматическогоДНК-секвенированияALFexpress II
СистемаавтоматическогоДНК-секвенированияALFexpress II
Для определения нуклеотидной последовательности ДНК используется система автоматического ДНК-секвенирования ALFexpress II (Amersham Pharmacia Biotech Ltd., USA) (рис. 1.17).
|
Современные аппаратура и методы исследования биологических систем. Большой практикум. Учебебное пособие |
56 |