

IV. Механизм Формирования патологии у плода при алкоголизме беременной женщины.
Алкоголь проходит через плаценту и попадает в организм плода. Вреден и сам спирт (этанол), и продукты его распада (ацетальдегид).
Спирт может вызывать спазм сосудов плаценты и пуповины, что приводит к кислородному голоданию плода.
Ацетальдегид: снижает уровень цинка в плодовых клетках, что нарушает их рост и развитие; обладает мутагенным эффектом; обусловливает дефицит витаминов. В итоге страдают многие органы и системы плода, но более всего - центральная нервная система.
Употребление алкоголя женщиной во время беременности значительно повышает риск невынашивания, мертворождения, рождения детей с синдром алкогольной фетопа-тии (аномалии развития сердца, наружных половых органов, нарушение функции центральной нервной системы, низкая масса тела при рождении, нарушения строения позвоночника, включая зрта Ъфс1аь отставание ребенка в физическом и умственном развитии). Последствия алкогольного поражения плода необратимы и практически не поддаются лечению.
Характер последствий алкогольного воздействия на плод зависит от очень многих причин. Безусловно, важнейшую роль играют количество спиртных напитков и частота их употребления. Ежедневный прием беременной 30 граммов спирта (других алкогольных напитков в пересчете на спирт), сопровождается высоким риском синдрома алкогольной фетопатии у будущего ребенка. Однако нередко этот синдром встречается у детей,
33
матери которых употребляли 3-5 граммов спирта ежедневно! Единой безопасной для всех дозы не существует!
V. Методы изучения наследственных болезней.
Клинико-генеалогический метод заключается в составлении родословной записи с последующим анализом проявления признака, характерного для конкретной наследственной болезни на протяжении возможно большего числа поколений родственников пациента.
Признаками наследственных болезней, установленных с помощью «родословных», являются:
обнаружение болезни «по вертикали»: из поколения в поколение беспрерывно (при доминантном типе наследования) или с некоторыми перерывами (при рецессивном типе наследования);
менделевские соотношения между, числом больных и здоровых сибсов (3:1; 1:1; 1:0);
большая частота заболевания среди родственников, чем среди неродственников.
Близнецовый метод состоит в сопоставлении внутрипарной конкордантности (идентичности) одно- и двуяйцевых близнецов, живущих $ разных и в одинаковых условиях, по анализируемому патологическому признаку.
В среднем на каждые 100 одноплодных родов приходятся одни близнецовые (многоплодные); при этом однояйцевые близнецы рождаются реже, чем двуяйцевые, примерно в 3-4 раза.
О наследственной природе патологии свидетельствует высокая конкордантность по анализируемому признаку однояйцевых близнецов, живущих в разных условиях, и, наоборот, низкая конкордантность двуяйцевых близнецов, особенно живущих в одинаковых условиях. Надротив, высокая конкордантность по какому-либо патологическому признаку одно- и двуяйцевых близнецов, живущих в одинаковой среде, явно говорит против наследственного происхождения данной патологии и, наоборот, подтверждает решающее значение в ее развитии экзогенных (внешних) факторов.
Популяционно-статистический метод заключается в составлении родословных среди большой группы населения, в пределах области или целой страны, в исследовании генетических изолятов. Изолят - это группа людей, от 500 человек до нескольких тысяч, живущая изолированно от всего остального населения страны. Генетический изолят характеризуется большей частотой эндогамных браков, что способствует увеличенитю количества случаев наследственных заболеваний в анализщируемой группе.
Цитологический метод - установление генетического пола при исследовании клеток на наличие телец Барра. Когда в клетке присутствует две X хромосомы (как у нормальной женщины), одна из них (тельце Барра) инактивируется и конденсируется на ядерной мембране. Отсутствие тельца Барра свидетельствует о наличии только одной X хромосомы (у нормального мужчины (ХУ) и при синдроме Шершевского-Тернера (ХО)). Тельца Барра наиболее легко определяются в мазках многослойного эпителия, которые получают путем соскабливания буккальной слизистой оболочки.
Биохимический и иммунологический методы заключаются в исследовании биохимических признаков, заведомо специфичных для определенных наследственных болезней. Так, например, для диагностики фенилпировиноградной олигофрении в мозе определяют фенилпировиноградную кислоту; для диагностики серповидно-клеточной анемии (8-гемоглобиноза) исследуют наличие в крови 8-гемоглобина; для выявления иммунодефи-цитных состояний определяют содержание различных антител и популяций лимфоцитов.
Дерматоглифический метод - выявление наследственных болезней по рисунку ладоней. В настоящее время используется крайне редко
34
Цитогенетический метод состоит в микроскопическом исследовании структуры и числа хромосом клеток (лейкоцитов, эпителия и др.). Изменение структуры и числа хромосом (хромосомные аберрации) является признаком наследственной природы болезни.
Молекулярно-генетический реализуется с помощью блот-гибридизации по Сау-зерну (введение флюоресцентной метки - ДНК-зонд) и амплификации (увеличении числа копий) участков ДНК при помощи ПЦР(полимеразной цепной реакции).
Метод ПЦР отличается очень высокой чувствительностью, позволяет обнаружить в пробе крайне малые концентрации ДНК. Тот же способ пригоден и для анализа следовых последовательностей РНК, для этого РНК переводят в последовательности комплементарной ДНК (кДНК), используя обратную транскриптазу. Метод получил широкое использование в пренатальнои диагностике наследственных болезней, выявлении вирусных инфекций, а также в судебной медицине.
VI. Мутагены. Классификация мутаций. Обозначения, принятые для изображения генеалогического дерева. Типы наследования дефектов в геноме. Мутация - скачкообразное изменение признака вследствие количественных или качественных изменений генотипа (мутагены -факторы, вызывающие мутации).
Мутации можно классифицировать, в зависимости от типа поврежденных клеток: соматические (не передаются по наследству, хотя и поражают генетический аппарат клетки) и гаметические (передаются по наследству); по степени совместимости с жизнью: летальные (сопровождаются гибелью организма внутриутробно, или сразу после рождения), сублетальные (гибель наступает до периода полового созревания), гипогенитальные (сопровождаются бесплодием); по характеру изменения генотипа в соответствии с тремя уровнями организации генетического материала: генные, хромосомные, геномные и цито-плазматические.
I, Генные мутации связаны с изменением структуры отдельных генов (участков ДНК, кодирующих синтез одного белка, одного признака);
Моногенная - мутация в одном гене с изменением одного признака (например, альбинизм, короткопалость); моногенные мутации обуславливают истинные наследственные заболевания. Вариантом моногенной является точковая мутации - повреждение (замена, выпадение, вставка) одного нуклеотида в гене, ведущая к нарушению аминокислотной последовательности в белке (ферментопатии, серповидно-клеточная анемия и др.).
Полигенные - одновременные мутации в различных генах различных хромосом, обуславливающие однбйаправленные изменения в организме, которые определяют предрасположенность к некоторым заболеваниям (см. «мультифакториальные болезни»)
П. Хромосомные мутации - структурные перестройки в отдельных хромосомах.
Делеция - это потеря части хромосомы в результате ее разрыва. Большинство деле-ций детальны в результате потери огромной части генетического материала. Делеция короткого плеча 4 хромосомы приводит к развитию синдрома Вольфа; делеция короткого плеча 5 хромосомы.- синдрома кошачьего крика (сп Ли ска1) - мяуканье и звуки подобные кошачьему крику типичны для этой патологии, часто наблюдается отставание в умственном развитии и пороки сердца.
Транслокация - это перенос отдельного сегмента одной хромосомы в другую хромосому. При сбаласнсированной транслокации весь генетический материал сохраняется и остается фунционально способным, поэтому фенотипических проявлений нет. У таких людей могут формироваться аномальные гаметы.
Дупликация - удвоение участка хромосомы.
Инверсии ^- поворот участка хромосомы на 180°.
35
III. Геномные мутации - изменения числа хромосом в наборе, не сопровождаемые изменением их структуры. Число хромосом при этом меняется некратно - формируется анеуплоидный набор хромосом. Кратное изменение числа хромосом {полиплоидия) несовместимо с жизнью.
1. Моносомии - уменьшение количества хромосом.
• Синдром Шерешевского-Тернера (яичниковая диегенезия) - моносомия половых хромосом, встречается довольно часто. Отсутствует одна Х-хромосома (45,ХО). В некоторых случаях вторая Х-хромосома присутствует, но в ней выявляютсятяжелые нарушения (изохромосома, частичная делеция и др.). Потеря второй X-хромосомы обычно приводит к гибели плода.
У выживших детей наблюдается лимфэдема шеи, которая присутствует и у взрослых, приводя к формированию толстой шеи. Часто наблюдаются врожденные аномалии сердца, низкий рост, ожирение и нарушения строения скелета. Интеллект не нарушен. В присутствии одной Х-хромосомы (и отсутствии У-хромосомы) примитивные половые железы развиваются как яичники. Яичники остаются маленькими и в них не обнаруживаются примордиальные фолликулы. Также нарушается синтез эстрогенов, что проявляется нарушением эндометриального цикла (аменоррея) и слабым развитием женских вторичных половых признаков. Диагноз может быть поставлен при отсутствии телец Барра в со-скобах буккального эпителия у лиц, имеющих женский фенотип или при анализе карио-типа,
• Аутосомные моносомии обычно летальны, поскольку теряется огромное количество генетического материала.
2. Триосомии - увеличение количества хромосом на одну.
• Синдром Кляйнфельтера (тестикулярная диегенезия) - трисомия половыххромосом - встречается довольно часто. Проявляется наличием лишней Х~хромосомы (47, XXV), реже больные с синдромом Кляйнфельтера могут иметь двеи более лишних Х-хромосом (48, XXXV или 49, ХХХХУ). Формируется мужскойфенотип.
До пубертатного периода никаких клинических проявлений не наблюдается. Лишняя Х-хромосома нарушает нормальное развитие яичек в пубертатном периоде неизвестным способом. Яички остаются маленькими и не продуцируют сперматозоиды, больные обычно бесплодны. Уровень тестостерона в крови низкий, что приводит к нарушению развития вторичных половых признаков. У больных имеется склонность к высокому росту (тестостерон ускоряет окостенение эпифизов) и евнухоидный внешний вид с высоким голосом, маленьким пенисом и ростом волос по женскому типу. Также иногда наблюдается гинекомастия. Иногда наблюдается снижение интеллекта. Диагноз синдрома Кляйнфельтера может быть установлен при нахождении телец Барра в соскобах буккального эпителия у лиц, имеющих мужской фенотип или при анализе кариотипа.
Синдром XXX ("суперженщины") - присутствие третьей Х-хромосомы у женщин. Большинство пациентов являются фенотипически здоровыми. У некоторых наблюдаются нарушения менструального цикла и снижение фертильности (плодовитости).
Синдром ХУУ - присутствие лишней У-хромосомы у мужчин. Большинство пациентов не имеют признаков какой-либо патологии. У некоторых может наблюдаться агрессивность поведения и легкое отставание в умственном развитии.
Синдром Дауна является наиболее частым аутосомным нарушением. Он возникает в результате наличия третьей 21 хромосомы, что приводит к развитию характерных клинических проявлений.
Дети имеют специфический косой разрез глаз с уплощенным профилем, косоглазие, резко выраженные вертикальные кожные складки, прикрывающие медиальный угол глазной щели, постоянным признаком является отставание в умственном развитии, 30% пациентов имеют врожденные пороки сердца. Наличие иммунодефицита у больных бу-
36
славливает высокую инфекционную и онкологическую (лейкозы) заболеваемость. Мужчины с синдромом Дауна обычно бесплодны, а женщины могут рожать детей, при этом вероятность рождения здорового ребенка составляет 50%, поскольку только половина гамет содержит лишнюю 21 хромосому.
• Синдром Эдвардса - трисомия 18 хромосомы (47ХХ/ХУ, +18) встречается редко.Клинически он проявляется отставанием в физическом и умственном развитии, сопровождаемым характерными физическими недостатками, такими как "стопа рокера" исжатые в кулаки руки с перекрещивающимися пальцами. В результате тяжелых поражений дети редко выживают более года.
• Синдром Патау - трисомия 13 хромосомы (47ХХ/ХУ, +13) также встречается редко. Большинство детей умирает сразу после рождения.
Трисомия 13 хромосомы характеризуется нарушением развития подкорковых структур мозга (отсутствие обонятельных луковиц, слияние лобных долей и единственный желудочек головного мозга) и срединных структур лица (расщепление губы, расщепление твердого неба, дефекты носа, единственный глаз [циклоп]).
IV, Цитоплазматические (митохондриальные) мутации возникают в результате мутаций в плазмогенах, находящихся в ДНК-содержащих клеточных органеллах - митохондриях. Некоторые варианты патологии (отдельные формы мужского бесплодия, некоторые виды близнецовости) могут быть обусловлены этими же причинами. Патологичекие признаки передаются потомству только от матери.
В стандартной родословной используются простые условные обозначения и правила (рис. 2.3.1.):
Мужчины всегда изображаются в виде квадратов, женщины - в виде окружностей.
Пациент, обратившийся к генетику для составления родословной - пробанд - обозначается стрелкой.
Графически изображаемые связи между членами родословной бывают только трех видов: "мужья-жены", "дети-родители" и "братья-сестры".
Супруги, братья и сестры (в т. ч. двоюродные и троюродные) всегда изображаются на одном горизонтальном уровне (т. е. в одном поколении). Разница в возрасте не играет никакой роли.
Дети пробанда изображаются на горизонтальном уровне ниже пробанда, а его родители - на горизонтальном уровне выше пробанда. То же самое относится к детям и родителям всех братьев и сестер пробанда.
Все поколения нумеруются сверху вниз римскими цифрами, а все индивидуумы в каждом поколении - слева направо арабскими цифрами. Это позволяет обозначить каждого человека личным идентификационным номером (например -111:15, что означает 15-й индивидуум в третьем поколении).
37
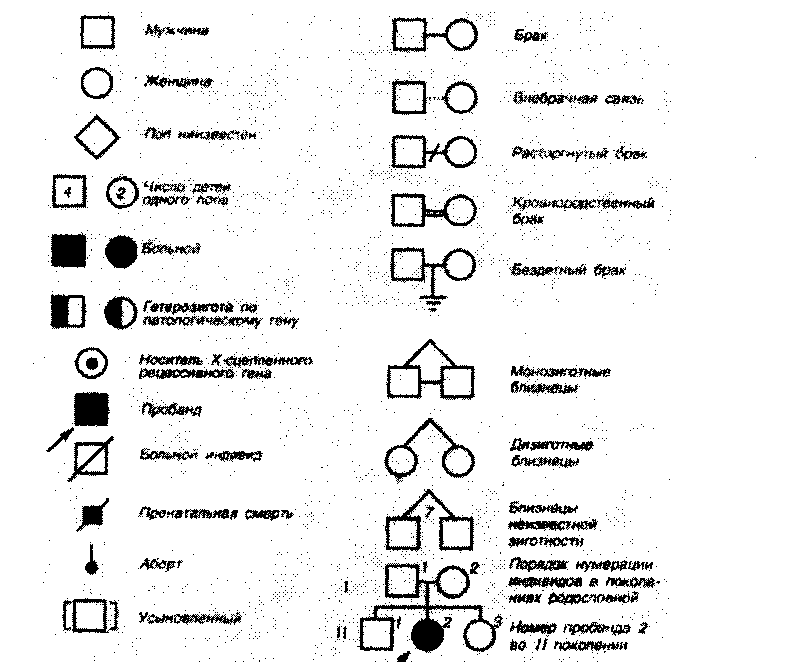
Рис. 2.3 Л. Символы, используемые при составлении родословной.
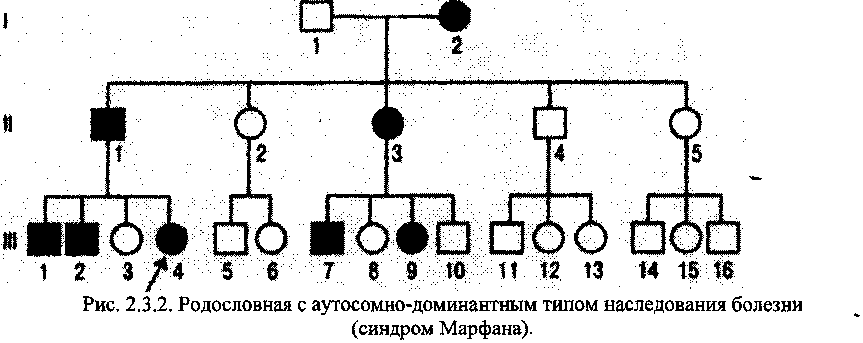
Типы наследования дефектов в геноме: Аутосомно-доминантный тип.
Критерии:
заболевание регулярно передается из поколения в поколение, т.е. прослеживается в родословной по вертикали,
риск рождения больного ребенка, если болен один из родителей, составляет 50%,
здоровые индивиды имеют здоровых потомков,
у больного индивида болен один из родителей,
оба пола поражаются с одинаковой частотой.
38
Примеры заболеваний: семейная гиперхолестеринемия, поликистоз почек (взрослый тип), наследственный эллиптоцитоз, наследственный сфероцитоз, болезнь Виллеб-ранда, семейный полипоз кишечника, ретинобластома, синдром Марфана, ахондроплазия (рис. 2.3.2.)
Аутосомно-рецессивный тип:
Критерии:
родители пробанда здоровы, но аналогичное заболевание может обнаруживаться у родных, двоюродных, троюродных сибсов, т.е. прослеживается в родословной по горизонтали (в одном поколении),
у больного родителя рождаются здоровые дети,
риск рождения больного ребенка равен 25%,
в случае кровнородственных браков наблюдается увеличение числа больных в родословной.
Примеры заболеваний: серповидноклеточная анемия, недостаточность миелоперокси-дазы, фенилкетонурия, альбинизм, болезнь Вильсона-Коновалова, галактоземия, гликоге-нозы, алкаптонурия
Х-сцепленный доминантный тип.
Критерии:
у больного пробанда обязательно болен один из родителей,
у больного отца все дочери больны, а сыновья здоровы,
у больной матери одинаковая вероятность рождения больной дочери и больного сына,
у здоровых родителей все дети будут здоровы,
больных женщин в 2 раза больше, чем больных мужчин.
Примеры заболеваний: гипофосфатемический рахит (витамин-Б-резистентный), псевдогиперпаратиреоидизм.
Х-сцепленный рецессивный тип.
Критерии:
заболевание наблюдается у мужчин-родственников пробанда по материнской линии,
сыновья никогда не наследуют заболевание отца,
у больного отца все его дочери здоровы и являются гетерозиготными носителями данного признака,
если женщина является гетерозиготньм носителем признака, то половина ее сыновей больны, а все дочери здоровы, причем половина ее дочерей является гетерозиготными носителями признака.
Примеры заболеваний: красно-зелёная цветовая слепота (дальтонизм), дефицит глюкозо-6-фосфатдегидрогеназы, мышечная дистрофия Дюшена, гемофилия А (дефицит VIII фактора), агаммаглобулинемия, тестикулярная феминизация.
У-сцепленный тип.
Критерии:
признак встречается только у лиц мужского пола,
признак передается от больного отца всем сыновьям.
Примеры заболеваний: некоторые формы гипертрихоза (избыточное оволосение), перепонки между пальцами.
Цитоплазматический (митохондриальный) тип.
Критерии:
признак встречается с одинаковой частотой у обоих полов;
признак передается от больной матери либо всему потомству, либо только его части в зависимости от попадания в зиготу аномальных плазмогенов от яйцеклетки.
Примеры заболеваний: одна из форм несращения остистых отростков позвонков, атрофия зрительного нерва Лебера, некоторые формы доброкачественных опухолей (он-коцитома).
39