

9.2.2. Хроматография на колонках
П ринцип
данного метода заключается в следующем.
В стеклянную колонку, заполненную
дисперсным носителем, пропитанным
неподвижной фазой, вносят раствор
анализируемой смеси и непрерывным
потоком подвижного растворителя элюируют
компоненты смеси, которые вымываются
из колонки в соответствии со своим
коэффициентом распределения. Титрованием
малых порций элюатов (или взвешиванием
содержащихся в них веществ) осуществляют
контроль за ходом элюации. В результате
последовательного извлечения компонента
смеси массовая доля его в элюате
возрастает, превышая максимальное
значение, затем снижается до нуля, что
указывает на конец элюации данного
компонента. При выделении следующего
компонента процесс повторяется.
ринцип
данного метода заключается в следующем.
В стеклянную колонку, заполненную
дисперсным носителем, пропитанным
неподвижной фазой, вносят раствор
анализируемой смеси и непрерывным
потоком подвижного растворителя элюируют
компоненты смеси, которые вымываются
из колонки в соответствии со своим
коэффициентом распределения. Титрованием
малых порций элюатов (или взвешиванием
содержащихся в них веществ) осуществляют
контроль за ходом элюации. В результате
последовательного извлечения компонента
смеси массовая доля его в элюате
возрастает, превышая максимальное
значение, затем снижается до нуля, что
указывает на конец элюации данного
компонента. При выделении следующего
компонента процесс повторяется.
Полученные результаты можно изобразить в виде хроматограммы, откладывая по оси абсцисс количество миллилитров элюата, по оси ординат массовую долю компонента в отдельных порциях элюата.
М
Рис.
9.2. Хроматограмма жирных кислот длительно
хранившегося подсолнечного масла:
1
– продукты окисления; 2
– неокисленные жирные кислоты
дуктов окисления жиров и фосфатидов
(рис. 9.2).
9.2.3. Газожидкостная хроматография
Метод газожидкостной хроматографии получил в настоящее время широкое распространение при исследовании ряда пищевых продуктов. Быстрому распространению этого метода способствуют его высокая разрешающая способность при разделении на компоненты сложных смесей близких по свойствам соединений, возможность более точного их количественного определения, простота препаративного выделения индивидуальных веществ и т. п.
Газожидкостная хроматография отличается от других методов распределительной хроматографии в основном тем, что в качестве подвижной фазы используется инертный газ (газ-носитель), а неподвижной фазой является жидкость, нанесенная на твердый носитель. Разделение смеси на индивидуальные вещества производят в специальных аппаратах – газовых хроматографах. Основными узлами газового хроматографа являются хроматографическая колонка, детектор и самописец. Необходимы для работы также источник газа – подвижная фаза.
В настоящее время в нашей стране и за
рубежом выпускаются удобные в работе
газовые хроматографы различных
конструкций, оснащенные компьютерами.
На рис. 9.3 изображена принципиальная
схема устройства газожидкостного
хроматографа.
настоящее время в нашей стране и за
рубежом выпускаются удобные в работе
газовые хроматографы различных
конструкций, оснащенные компьютерами.
На рис. 9.3 изображена принципиальная
схема устройства газожидкостного
хроматографа.
Рис. 9.3. Принцип устройства газожидкостного хроматографа: 1 – баллон с газом; 2 – редукционный клапан; 3 – вентиль тонкой регулировки; 4 – манометр; 5 – ротаметр; 6 – хроматографическая колонка; 7 – термостат для колонки; 8 – ввод анализируемой пробы; 9 – детектор; 10 – пленочный расходомер газа; 11 – регуляторы термостата; 12 – регуляторы-детекторы; 13 – регуляторы ос- новной линии самописца; 14 – регулятор чувствительности; 15 – панель для контрольных приборов; 16 – самописец; 17 – хроматограмма
Сущность метода газожидкостной хроматографии заключается в том, что хроматографическую колонку заполняют твердым порошкообразным носителем, пропитанным нелетучей жидкостью – неподвижной фазой. Колонку, помещенную в термостат, нагревают, одновременно пропуская через нее с постоянной скоростью инертный газ. При достижении определенной температуры в колонку с помощью микрошприца вводят раствор анали-зируемой смеси веществ, которые под влиянием повышенной температуры быстро превращаются в пар. Часть парообразных компонентов смеси, перемещаясь с непрерывно пропускаемым инертным газом вдоль колонки, растворяются в неподвижной фазе, другие увлекаются газом дальше вдоль колонки. Чем ниже растворимость парообразного компонента в жидкой фазе, тем быстрее он проходит через колонку.
Скорость продвижения отдельных компонентов смеси определяется коэффициентом распределения между газообразной и жидкой фазами:
К =
![]() = const,
(9.1)
= const,
(9.1)
где К – коэффициент распределения компонента; Сж – концентрация компонента в жидкой фазе; Сг – концентрация компонента в газовой фазе.
Поток выходящего из колонки газа-носителя последовательно выносит отдельные компоненты смеси, разделенные объемами чистого газа-носителя.
Изменение физических сил или химических свойств выходящего газа фиксируется детектором, сигнал которого усиливается и записывается на ленте самописца в виде хроматограммы (рис. 9.4). Обсчитывает результаты разделения вычислительная машина, которая часто входит в комплект хроматографа.
Р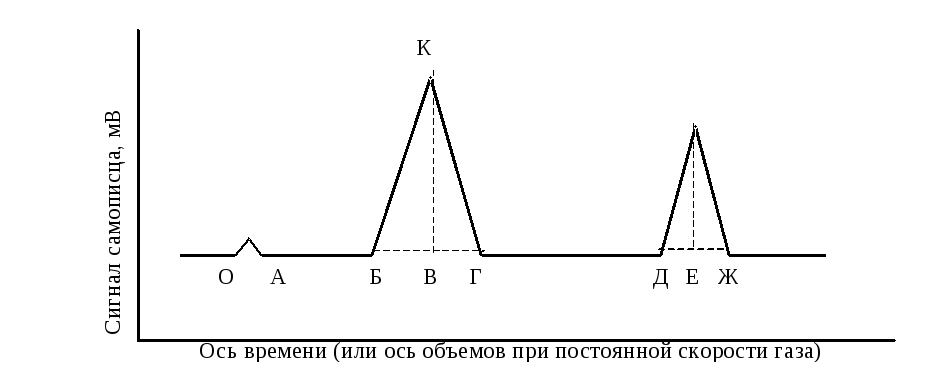 ис.
9.4. Пример хроматограммы
ис.
9.4. Пример хроматограммы
В некоторый период времени О проба вводится в колонку. До выхода отдельных компонентов пробы перо самописца вычерчивает прямую линию (ОА, АБ, ГД), называемую основной. В период времени А из колонки выходит газ-носитель. Объем газа, прошедшего через колонку за время ОА, представляет собой объем задержки колонки.
Разделяемые в колонке компоненты
движутся медленнее газа-носи-теля, и
первый компонент выходит из колонки в
период времени от О до Б. Интервал времени
между вводом пробы и появлением максимума
пика (ОВ) называют временем удерживания
данного компонента, а объем
газа-ностителя, прошедшего через колонку
за это время, – удерживаемым объемом
VR. Удерживаемый объем
является специфической характеристикой
данного компонента. Однако на практике
чаще всего используют понятие
относительного удерживаемого объема
V![]() ,
который вычисляют по формуле
,
который вычисляют по формуле
V![]() =
=
![]() ,
(9.2)
,
(9.2)
где VR – удерживаемый объем данного компонента; VRст – удерживаемый объем вещества, принятого за стандарт, измеренный при тех же условиях и на той же колонке.
Разделение смеси веществ характеризуется двумя независимыми факторами: коэффициентом разделения и эффективностью колонки. Коэффициент разделения определяет относительное положение пиков на хроматограмме, а эффективность колонки – форму пиков.
Коэффициент разделения вычисляют по формуле
=
![]() ,
(9.3)
,
(9.3)
где (VR)2 и (VR)1 – удерживаемые объемы компонентов 1 и 2.
При = 1 компоненты образуют неразделяемые пики. Коэффициент разделения можно изменять соответствующим подбором неподвижной фазы или температуры колонки. Хорошее разделение компонентов получается при коэффициенте разделения свыше 1,2. Учитывая ширину пиков и величину удерживаемых объемов двух компонентов, можно рассчитать степень разделения М этих компонентов по уравнению
М =
![]() ,
(9.4)
,
(9.4)
где Δу – разность удерживаемых объемов компонентов; у1 и у2 – ширина пиков компонентов 1 и 2.
Эффективность колонки измеряется числом теоретических тарелок. Согласно концепции «теоретических тарелок» хроматографическую колонку мысленно рассматривают как аппарат, состоящий из ряда элементов, называемых теоретическими тарелками. В каждом элементе (тарелке) при прохождении газа устанавливается полное равновесие между подвижной фазой – газом и неподвижной – жидкостью, нанесенной на твердый носитель. Размер теоретической тарелки зависит от коэффициентов распределения компонентов. В действительности, однако, хроматографический процесс протекает непрерывно во всех тарелках и полное равновесие в какой-либо точке не устанавливается. Поэтому практически можно рассчитать лишь высоту колонки, в которой происходит разделение, эквивалентное одной теоретической тарелке. Эта величина называется высотой, эквивалентной теоретической тарелке (ВЭТТ). Ее вычисляют делением длины колонки на число теоретических тарелок n, которое можно рассчитать по формуле
n
= 16![]() ,
(9.5)
,
(9.5)
где х – расстояние на основной линии между точкой ввода пробы и перпендикуляром, опущенным из точки максимума пика (см. рис. 9.4 – величина ОВ); у – ширина пика, определяемая касательными к точкам перегиба (см. рис. 9.4 – величина БГ)
Число теоретических тарелок указывает, сколько раз за время продвижения газа через колонку устанавливается равновесие между подвижной и неподвижной фазами. Оно является сложной функцией скорости потока газа, природы неподвижной фазы, свойств разделяемых компонентов, плотности заполнения колонки, температуры и других условий. Чем больше число теоретических тарелок, тем выше эффективность колонки и острее пики на хроматограмме.
Эффективность хроматографического анализа зависит от большого числа параметров. К числу таких параметров относятся: свойства растворителя – неподвижная фаза, температура опыта, длина колонки, ее форма и сечение, плотность заполнения колонки носителем; свойства газа-носителя, скорость и давление газового потока; количество нанесенного вещества и способ нанесения пробы, чувствительность детектора и стабильность его работы. При этом свойства и количество растворителя, длина и температура колонки определяют расстояние между пиками, а свойства, скорость и давление газа-носителя, плотность заполнения колонки, ее форма и сечение – размывание пика веществ, т. е. ширину пика.
Хроматографические колонки. В зависимости от конструкции газового хроматографа колонки бывают различной формы (прямые, U-образ-ные, спиральные) и изготавливаются из различных материалов (стекла, меди, латуни и др.). Диаметр колонок обычно составляет 4…8 мм; колонки с большим диаметром (20 мм) применяют только для препаративных целей. Длину колонки подбирают эмпирическим путем в зависимости от состава анализируемой смеси. Обычно на колонках длиной 2…3 м можно достичь удовлетворительного разделения. Более длинные колонки используют для работы с трудно разделяемыми смесями.
В связи с разработкой очень чувствительных детекторов в последнее время большое распространение получили капиллярные колонки, изготавливаемые из стекла или металла, с внутренним диаметром 0,01…0,1 см и длиной 15…60 м и более. На стенки капилляра наносят тонкий слой жидкости (3…5 мкм), выбранной в качестве неподвижной фазы. Носителем неподвижной фазы служат стенки капиллярной трубки. Преимуществом таких колонок является очень высокая эффективность и быстрота разделения пробы.
Неподвижная фаза. Свойства неподвижной фазы являются одним из важнейших факторов, влияющих на разделение смеси на компоненты. Выбор неподвижной фазы определяется, прежде всего, свойствами отдельных компонентов исследуемой смеси, задачей исследований и температурой опыта. Выбранная фаза должна обладать полной химической инертностью, малой вязкостью и быть практически нелетучей жидкостью при температуре опыта.
Жидкие фазы, наиболее употребимые при анализе смесей жирных кислот, спиртов, глицеридов и т. д., приведены в табл. 9.1.
При анализе очень сложных смесей используют несколько жидких фаз, применяя смесь жидкостей на одном носителе или последовательно соединенные колонки с различными жидкостями.
Носители неподвижной фазы. Выбранной жидкостью пропитывают твердый носитель. Носитель должен иметь большую удельную поверхность с широкими порами, достаточную механическую прочность быть инертным по отношению к анализируемым веществам и газу-носителю. В качестве носителей чаще всего применяют цеолит 545 (сорт кизельгура), огнеупорные кирпичи С-22 и стерхамол, хромосорбы «W» и «Р», а из отечественных – инзенский кирпич ИНЗ-600, диатомит и каолин.
Цеолит 545 и хромосорбы требуют предварительной дезактивации концентрированной соляной кислотой.
Для этого отобранную фракцию носителя помещают в стакан и заливают концентрированной соляной кислотой в таком количестве, чтобы носитель был весь покрыт жидкостью. После перемешивания (вращательными движениями, чтобы не измельчить носитель) смесь отстаивают в течение 1 ч. Сливают избыток кислоты и промывают носитель: вначале несколько раз водой (декантация), затем раствором 1 н. NаОН и еще раз водой до нейтральной реакции. После просушивания носителя в сушильном шкафу при температуре 145 °С до полного удаления воды его просеивают через сито с требуемым числом отверстий.
Таблица 9.1