


152 Antithrombotics
Intra-arterial Thrombus Formation (A)
Activation of platelets, e.g., upon contact with collagen of the extracellular matrix after injury to the vascular wall, constitutes the immediate and decisive step in initiating the process of primary hemostasis, i.e., cessation of bleeding. However in the absence of vascular injury, platelets can be activated as a result of damage to the endothelial cell lining of blood vessels. Among the multiple functions of the endothelium, the production of prostacyclin and nitric oxide (NO) plays an important role because both substances inhibit the tendency of platelets to adhere to the endothelial surface. Impairment of endothelial function, e.g., due to chronic hypertension, chronic elevation of plasma LDL levels or of blood glucose, and cigarette smoking, increases the probability of adhesion between thrombocytes and endothelium. The deceleration of fast flowing platelets occurs through an interaction between the glycoprotein Ibα (GP I) in the platelet membrane and von Willebrand factor in the endothelium and basal membrane (denuded after endothelial injury). For the proper activation of the platelet, interaction with subendothelial collagen of an additional platelet glycoprotein (GP IV) is necessary. As soon as platelets are activated (see p.154), they change their shape and gain af nity for fibrinogen. Thisresultsfromaconformational change of glycoprotein IIb/IIIa in the platelet membrane. Platelets can now be linked to each other via fibrinogen bridges (A).
Platelet aggregation proceeds like an avalanche because, once activated, one platelet can activate other platelets. On the injured endothelial cell a thrombus is formed, which obstructs blood flow. Ultimately, the vascular lumen is occluded by the thrombusasthe latter is solidified by vasoconstriction promoted by the release of serotonin and thromboxane A2 from the aggregated platelets and by locally activated thrombin. Thrombin plays a twofold part in thrombus
formation: as a protease, thrombin cleaves fibrinogen and thus initiates the formation of fibrin clot (blood coagulation, p.144). The effects of thrombin on platelets and endothelial cells, however, involve a proteolytic activation of receptorscoupled to G-proteins (so-called protease-activated receptors). When these events occur in a larger, functionally important artery, myocardial infarction or stroke may be the result.
Von Willebrand factor plays a key role in thrombogenesis. Lack of this factor is the cause of thrombasthenia, the inability to staunch bleeding by platelet aggregation. A relative deficiency of von Willebrand factor can be transiently relieved by injection of the vasopressin analogue desmopressin, because this substance makes factor available from stored supplies.
Formation, Activation, and Aggregation of Platelets (B)
Platelets are fragments of multicellular megakaryocytes. They constitute the smallest formed elements of blood (diameter 1–4 µm) and, devoid of a cell nucleus, are no longer capable of protein synthesis. Platelets can be activated by various stimuli, leading to:
Change in shape
Conversion of integrin GP IIb/IIIa into its active conformation
Release of active substances such as serotonin, platelet-activating factor (PAF), ADP, and thromboxane A2. All these substances activate other platelets.

Intra-arterial Thrombus Formation |
153 |
A. Thrombogenesis |
|
|
|
|
|
1. Adhesion |
2. Activation |
3. Aggregation |
|||
|
Endothelial defect |
|
|
||
Platelet |
Activated |
von Willebrand |
Collagen |
Fibrinogen |
|
not activated |
platelet |
factor |
|||
|
|
||||
B. Aggregation of platelets by the integrin GPIIb/IIIa |
|
Megakaryocyte |
|
|
Contact with |
|
collagen |
Activation |
ADP |
Thrombin |
|
|
Thromboxane A2 |
|
Serotonin |
Glyco- |
|
protein |
|
IIb/IIIa |
|
Platelet |
|
|
Platelet |
Fibrinogen |
Glycoprotein |
|
IIb/IIIa |
|
Fibrinogen- |
|
binding: |
|
impossible |
Aggregation |
possible |

154 Antithrombotics
Inhibitors of Platelet Aggregation (A)
Collagen, thrombin, ADP, and thromboxane A2 are the most important mediators that induce maximal activation and aggregation of platelets. The first essential step in platelet activation is mediated by direct contact with collagen, which can bind to different proteins in the platelet membrane. The most important “collagen receptor” in the platelet membrane is glycoprotein VI (GP VI). Activation induces a change in platelet shape and triggers secretion of substances stored in intracellular platelet granula (e.g., ADP, serotonin). In addition, GP VI stimulates cyclooxygenase (COX-1), causing thromboxane A2 to be produced and released from arachidonic acid (p.196).
The propensity of platelets to aggregate can be inhibited by various pharmacological interventions.
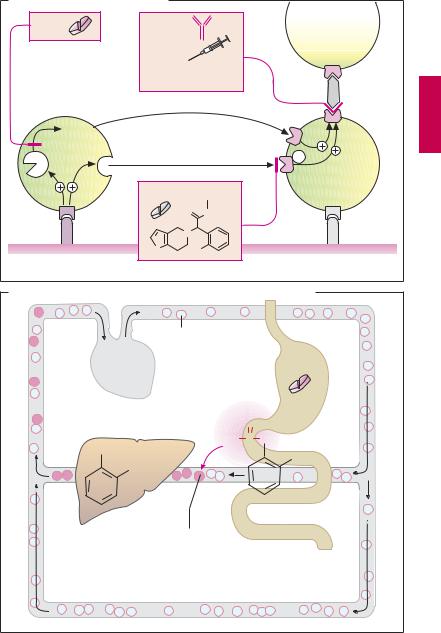
Acetylsalicylic acid (ASA) prevents COX- 1-mediated synthesis of thromboxane. Low daily doses (75–100 mg) may be suf cient. Indications include prophylaxis of re-infarc- tion after myocardial infarction and of stroke. Despite the low dosage, adverse effects such as gastric mucosal damage or provocation of asthma attacks cannot be ruled out.
Available alternatives to ASA are the ADP receptor antagonists ticlopidine and clopidrogel, which can also be given orally. Similarly to ASA, ticlopidine and clopidrogel cause an irreversible inhibition of platelet function. Both substances are inactive precursors that are converted by hepatic cytochrome P450 to an active metabolite that binds covalently to a subtype (P2Y12) of ADP receptors on platelets. Consequently, ADP-mediated platelet aggregation is inhibited for the duration of the platelet life cycle (~ 7–10 days). Ticlopidine may cause serious adverse effects, including neutropenia and thrombopenia. The successor substance, clopidrogel, is better tolerated.
Antagonists at the integrin glycoprotein IIb/IIIa. Available agents are suitable only for parenteral administration and, in clinical settings, are used in percutaneous coronary balloon distension or in unstable angina pectoris. They block the fibrinogen cross-linking protein and thus decrease fibrinogen-medi- ated meshing of platelets independently of the precipitating cause. Abciximab is a chimeric Fab-antibody fragment directed against GP IIb/IIIa protein. Tirofiban and eptifibatide act as competitive antagonists at the fibrinogen binding site. Because abciximab adheres to GPIIb/IIIa for a long time, 24–48 hours are required after injection of the drug before platelet aggregation again becomes possible. The effects of eptifibatide and tirofiban dissipate within a few hours. Because GP IIb/IIIa antagonists inhibit the common final pathway in platelet activation, they pose a risk of bleeding during treatment.
Presystemic Effect of ASA
The inhibition of platelet aggregation by ASA results from acetylation and blockade of platelet COX-1 (B). The specificity of this reaction is achieved in the following manner: irreversible acetylation of the enzyme already occurs in the blood of the splanchnic region, that is, before the liver is reached. Since ASAissubjecttoextensive presystemic deacetylation,cyclooxygenases located posthepatically (e.g., in endothelial cells) are hardly affected. Confinement of COX-1 inhibition to platelets is further accentuated because enzyme can be re-synthesized in normal cells having a nucleus but not in the anuclear platelets.
Inhibitors of Platelet Aggregation |
155 |
A. Inhibitors of platelet aggregation |
|
|
||
ASA |
|
Abciximab |
|
|
|
|
|
|
|
Acetylsalicylic acid |
Eptifibatide, |
|
||
|
|
a peptide |
|
|
|
|
Tirofiban, |
|
|
|
|
nonpeptide |
|
Fibrinogen |
|
|
|
|
|
|
|
GPIIb/IIIa antagonists |
||
|
|
|
|
GPIIb/IIIa |
|
Throm- |
|
|
TP |
|
boxane A2 |
|
|
|
Cox1 |
Secretion |
ADP |
Gi |
|
|
|
|
|
P2Y12 |
|
|
Clopidogrel |
|
CH3 |
|
|
|
|
|
GPVI |
|
O |
|
O |
|
|
|
|
|
|
|
N |
|
|
|
|
S |
Cl |
|
Collagen |
|
ADP-receptor antagonists |
||
|
|
|||
B. Presystemic inhibition of platelet aggregation by acetylsalicylic acid |
||
Platelets |
|
|
|
Low dose |
|
O |
Acetylsalicylic |
|
H3C C O |
acid |
|
|
||
OH |
COOH |
|
COOH |
||
|
||
Acetylation of |
|
|
COX in platelets |
|
|