


26 Distribution in the Body
Membrane Permeation
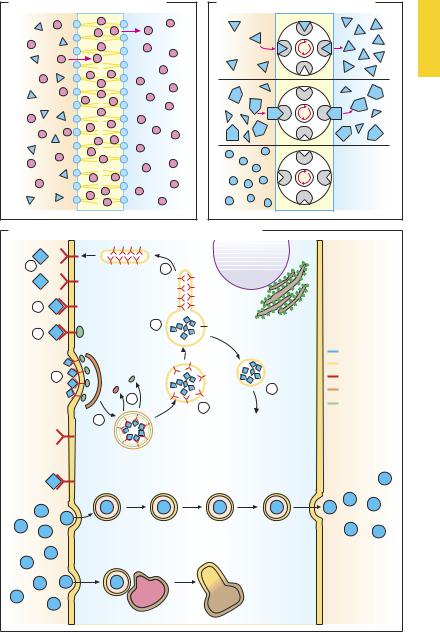
An ability to penetrate lipid bilayers is a prerequisite for the absorption of drugs, their entry into cell or cellular organelles, and passage across the blood–brain barrier. Owing to their amphiphilic nature, phospholipids form bilayers possessing a hydrophilic surface and a hydrophobic interior (p.20). Substances may traverse this membrane in three different ways.
Diffusion (A). Lipophilic substances (red dots) may enter the membrane from the extracellular space (area shown in ochre), accumulate in the membrane, and exit into the cytosol (blue area). Direction and speed of permeation depend on the relative concentrations in the fluid phases and the membrane. The steeper the gradient (concentration difference), the more drug will be diffusing per unit of time (Fick’s law). The lipid membrane represents an almost insurmountable obstacle for hydrophilic substances (blue triangles).
Transport (B). Some drugs may penetrate membrane barriers with the help of transport systems (carriers), irrespective of their physicochemical properties, especially lipophilicity. As a prerequisite, the drug must have af nity for the carrier (blue triangle matching recess on “transport system”) and, when bound to the carrier, be capable of being ferried across the membrane. Membrane passage via transport mechanisms is subject to competitive inhibition by another substance possessing similar af nity for the carrier. Substances lacking in af nity (blue circles) are not transported. Drugs utilize carriers for physiological substances: e.g., L-dopa uptake by L-amino acid carrier across the blood–intestine and blood–brain barriers (p.188); uptake of aminoglycosides by the carrier transporting basic polypeptides through the luminal membrane of kidney tubularcells(p.280). Onlydrugsbearingsuf-
ficient resemblance to the physiological substrate of a carrier will exhibit af nity to it.
The distribution of drugs in the body can be greatly changed by transport glycoproteins that are capable of moving substances out of cells against concentration gradients. The energy needed is produced by hydrolysis of ATP. These P-glycoproteins occur in the blood–brain-barrier, intestinal epithelia, and tumor cells, and in pathogens (e.g., malarial plasmodia). On the one hand, they function to protect cells from xenobiotics; on the other, they can cause drug resistance by preventing drugs from reaching effective concentrations at intracellular sites of action.
Transcytosis (vesicular transport, C). When new vesicles are pinched off, substances dissolved in the extracellular fluid are engulfed and then ferried through the cytoplasm, unless the vesicles (phagosomes) undergo fusion with lysosomes to form phagolysosomes and the transported substance is metabolized.
Receptor-mediated endocytosis (C). The drug first binds to membrane surface receptors (1, 2) whose cytosolic domains contact special proteins (adaptins, 3). Drug–receptor complexes migrate laterally in the membrane and aggregate with other complexes by a clathrin-dependent process (4). The affected membrane region invaginates and eventually pinches off to form a detached vesicle (5). The clathrin and adaptin coats are shed (6), resulting in formation of the “early” endosome (7). Inside this, proton concentration rises and causes the drug–re- ceptor complex to dissociate. Next, the re- ceptor-bearing membrane portions separate from the endosome (8). These membrane sections recirculate to the plasmalemma (9), while the endosome is delivered to the target organelles (10).
Membrane Permeation |
27 |
A. Membrane permeation: diffusion |
B. Membrane permeation: transport |
C. Membrane permeation: vesicular uptake, and transport |
|
||
1 |
9 |
|
|
|
|
|
|
2 |
|
|
|
3 |
8 |
pH 5 |
|
|
|
|
|
|
|
|
Ligand |
|
|
|
Plasmalemma |
4 |
|
|
Receptor |
|
|
10 |
Clathrin |
|
6 |
7 |
Adaptins |
|
|
|
|
|
5 |
|
|
|
Vesicular transport |
|
|
|
Lysosome |
Phagolysosome |
|
Extracellular |
Intracellular |
Extracellular |
|
28 Distribution in the Body
Possible Modes of Drug Distribution
Following its uptake into the body, the drug is distributed in the blood (1) and through it to the various tissues of the body. Distribution may be restricted to the extracellular space (plasma volume plus interstitial space) (2) or may also extend into the intracellular space (3). Certain drugs may bind strongly to tissue structures so that plasma concentrations fall significantly even before elimination has begun (4).
After being distributed in blood, macromolecular substances remain largely confined to the vascular space, because their permeation through the blood–tissue barrier, or endothelium, is impeded, even where capillaries are fenestrated. This property is exploited therapeutically when loss of blood necessitates refilling of the vascular bed, for instance by infusion of dextran solutions (p.156). The vascular space is, moreover, predominantly occupied by substances bound with high af nity to plasma proteins (p.30; determination of the plasma volume with protein-bound dyes). Unbound, free drug may leave the bloodstream, albeit with varying ease, because the blood–tissue barrier (p.24) is differently developed in different segments of the vascular tree. These regional differences are not illustrated in the accompanying figures.
Distribution in the body is determined by the ability to penetrate membranous barriers (p.20). Hydrophilic substances (e.g., inulin) are neither taken up into cells nor bound to cell surface structures and can thus be used to determine the extracellular fluid volume (2). Lipophilic substances diffuse through the cell membrane and, as a result, achieve a uniform distribution in body fluids (3).
Body weightmaybe broken down as illustrated in the pie-chart. Further subdivisions are shown in the panel opposite.
The volume ratio of interstitial: intracellular water varies with age and body weight. On a percentage basis, interstitial fluid vol-
Solid substances and structurally bound water
40% |
20% |
40% |
Intracellular water |
Extracellular water |
|
and erythrocytes |
ume is large in premature or normal neonates (up to 50% of body water), and smaller in the obese and the aged.
The concentration (c) of a solution corresponds to the amount (D) of substance dissolved in a volume (V); thus, c = D/V. If the dose of drug (D) and its plasma concentration (c) are known, a volume of distribution (V) can be calculated from V = D/c. However, this represents an apparent (notional) volume of distribution (Vapp), because an even distribution in the body is assumed in its calculation. Homogeneous distribution will not occur if drugs are bound to cell membranes (5) or to membranes of intracellular organelles(6)orarestoredwithinorganelles (7). In these cases, plasma concentration c becomes small and Vapp can exceed the actual size of the available fluid volume. Conversely, if a major fraction of drug molecules is bound to plasma proteins, c becomes large andthecalculatedvalueforVapp maythenbe smaller than that attained biologically.
Possible Modes of Drug Distribution |
29 |
|||
|
|
|
|
|
A. Compartments for drug distribution |
|
|
||
1 |
|
2 |
3 |
4 |
Intravascular |
Intravascular and |
Extraand |
Tissue binding |
|
|
|
interstitial |
intracellular |
|
Plasma |
Interstitium |
|
|
|
6% |
|
25% |
|
|
4% |
|
65% |
|
|
|
|
|
|
|
Erythrocytes |
|
Aqueous spaces of the organism |
||
Intracellular space |
|
|
||
|
|
|
|
Lysosomes |
|
|
|
|
Mito- |
|
|
|
|
chondria |
|
|
|
|
Nucleus |
|
|
|
|
Cell |
|
|
|
|
membrane |
5 |
|
6 |
|
7 |