

Контрольные вопросы и примеры к III главе
Что такое газовая смесь?
Дать формулировку закона Дальтона.
Что называется парциальным давлением?
Что называется массовой, объёмной и мольной долями?
Что называется парциальным, или.приведенным, объемом?
Какая существует зависимость между удельным объемом, плотностью, молекулярной массой и газовой постоянной?
Почему молекулярная масса смеси называется средней молекулярной массой?
Как производится пересчет массового состава в объемный и объемного в массовый?
-9. -Как определяется газовая постоянная смеси по массовым и объемным долям?
- 10. Как определяется парциальное давление газа в смеси по массовым и объемным долям?
11. Как определяется средняя молекулярная масса смеси газов? Пример 3-1. Определить среднюю молекулярную массу сухого атмосферного воздуха, если принять, что он состоит по объему из 21% 02 и 79% N2.
По уравнению (3-9) имеем
Р = 'iPi + r2\i2 = 32-0,210 + 28,016-0,79 = 28,93.
Пример 3-2. Определить газовую постоянную, плотность и парциальные давления для смеси, состоящей из 20 массовых долей воздуха и одной массовой доли светильного газа. Плотность светильного газа при температуре 273Q К и давлении 101325 н1м%-равна 0,52 кг/ж3.
Газовую постоянную светильного газа определяем из уравнения ' Клапейрона:
R=
101325
=714 дж/(кг-град).
/
0,52-273 к
'
Газовая постоянная воздуха 287,04 дж/(кг-град).
Газовую постоянную смеси газов определяем по уравнению (3-3): # = ЯЛ + £2#2 = 287,04-20/21 + 714-1/21 = 306,3 дж/(кг-град). Плотность смеси определяем по уравнению Клапейрона: р = р1ЯТ = 101325 : 306,30-273 = 1,21 кг/м3. Парциальное давление воздуха находим по уравнению (3-11):
р^ра= 101325- — =90 100 н]м\
1 кв1 Я ■ 21 306,3
Парциальное давление светильного газа равно
Р^ = Р§2~ = 101325— ^—=11225 н/м2
™ ^&2 Я 21 306,30
Глава IV реальные газы
§ 4-1. Свойства реальных газов
. Реальные газы отличаются от идеальных газов тем, что молекулы этих газов имеют конечные собственные объемы и связаны между собой силами взаимодействия,, которые имеют электромагнитную и квантовую природу. Эти силы существуют между любыми молекулами при любых условиях и уменьшаются с увеличением расстояния между молекулами. При сближении молекул на малые расстояния силы притяжения резко уменьшаются и пере^
гающие очень- больших значений. °
Из-за наличия сил взаимодействия между молекулами и конечности их объема законы идеальных газов ни при каких условиях не могут~быть строго применимы к реальным газам.
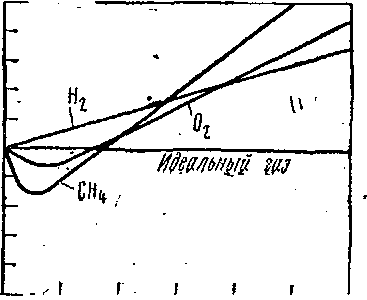
При практических расчётах различных свойств реальных газов находит широкое применение величина
отношения рь/ЯТ = С, которая по-
лучила название коэффициента ежи- О Ш ЮО Ш Щ 1Шр/ар маемости. (Эта величина не является коэффициентом термодинамического - Рис. 4-1
сжатия, который рассматривается в § 4-3).
Так как.для идеальных газов при любых условиях ро = НТ и С = = 1, то величина коэффициента сжимаемости выражает отклонение свойств реального газа от свойств идеального. Величина С для реальных газов в зависимости от давления и температуры может принимать значения больше и меньше единицы и только при очень малых давлениях и высоких температурах она практически равна единице.
На рис. 4-1 показана зависимость величины С от давления при температуре г* = 0° для некоторых газов. Повышение давления и понижение температуры, увеличение концентрации молекул газа и уменьшение расстояния между ними, усиливает отклонения свойств реального от свойств идеального газа. Из уравнения Клапейрона — Менделеева следует, что при любой постоянной температуре зависимость ри от р должна изображаться прямой, параллельной оси давления. В действительности изотермы всех газов представляют собой кривые даже в области не очень высокихдавлений, а при давлениях от 200 бар и выше кривые довольно круто поднимаются вверх.
*
Здесь р0
и и0
— давление и удельный вес воздуха при
нормальных ■физических условиях, а
отношение ри/р0«0
— так называемое число Амага.
В. В. Нащокин
33
— const. Из этого следует, что при малых р и больших' v произведение pv изменяется очень мало и остается, почти постоянным. Следовательно, чем больше разрежение, тем с большей точностью удовлетворяется уравнение Клапейрона — Менделеева для любого реального газа.
 Температура,
соответствующая изотерме с точкой
минимума на оси
ординат (р
= 0),
называется температурой
Бойля. Изотерма,
начи-
нающаяся в точке Бойля, на
некотором протяжении будет
прямой,
параллельной оси абсцисс, т.
е. здесь точно соблюдается закон
pv)poV0
=
const.
Все изотермы, начинающиеся выше
температуры Бойля,
т~>п
-,у <* имеют
вид восходящих кривых. Для воз-
Температура,
соответствующая изотерме с точкой
минимума на оси
ординат (р
= 0),
называется температурой
Бойля. Изотерма,
начи-
нающаяся в точке Бойля, на
некотором протяжении будет
прямой,
параллельной оси абсцисс, т.
е. здесь точно соблюдается закон
pv)poV0
=
const.
Все изотермы, начинающиеся выше
температуры Бойля,
т~>п
-,у <* имеют
вид восходящих кривых. Для воз-
1,8
1,6
1,2
1,0 0,8 0,6 0,4
о,г
Таким образом, свойства реальных газов как в количественном, так и качественном отношениях'значительно отличаются от свойств идеальных газов. Поэтому все результаты, полученные для реальных газов на основе законов идеальных газов, нужно рассматривать как приближенные и справедливые при очень больших разрежениях (р ->- 0).
Отличие свойств любого реального газа от свойств идеального заставило ученых разрабатывать новые уравнения состояния, которые связывали бы значения р, и и Т и давали бы возможность рассчитывать некоторые свойства газов для разных условий, не прибегая к дорогостоящим, не всегда доступным прямым измерениям.
уравнение'
pv
= RT
V+l t!V
В этом уравнении коэффициенты Вч при степенях \1о в правой части уравнения, называемые еириальными коэффициентами, выражаются через потенциальную энергию взаимодействия молекул данного газа и температуру Т, а и = 2, 3, 4, ... п — порядковый номер ви-риального коэффициента.
Однако^полученное уравнение в общем виде не может быть использовано для непосредственных расчетов реальных газов.
*
Уравнение получено Боголюбовым и
Майером и поэтому носит их имя.
11в зависимости от расстояния между ними (так называемая потенциальная кривая) и при наличии определенного количества экспериментальных данных, может быть получено расчетное уравнение того или '|иного реального газа в довольно широком диапазоне изменения параметров. Из-за сложности вычисления вириальных коэффициентов Добычно ограничиваются расчетом первых двух из них. Тогда расчетное уравнение имеет такой вид:
где А я В — первый и второй вириальные коэффициенты, являющиеся функцией только температуры.
В настоящее время уравнения подобного вида получили широкое распространение при расчете свойств многих реальных газов.
Наиболее простым и качественно верно отражающим поведение ■V реального газа, я'вляется уравнение Ван-дер-Ваальса, которое получается как частный случай из общего уравнения состояния Майера — Боголюбова, если пренебречь в правой части всеми членами, одержавшими 1/у во второй степени и выше.
§ 4-2. Уравнение состояния Ван-дер-Ваальса
, Уравнение состояния Ван-дер-Ваальса является одной из первых попыток аналитически описать свойства реальных газов. Это уравне-у ние наглядно показывает качественные особенности реальных газов и их отличие от идеальных.
Как уже отмечалось, реальные газы отличаются от идеальных наличием сил взаимодействия между молекулами и объемом самих молекул. Силы взаимодействия очень велики у твердых и жидких тел и. достаточно велики у газов, близких к переходу от газообразного в жидкое состояние.
. Следовательно, чем дальше состояние газа находится от области пе-.рехода в жидкость и чем больше расстояние между молекулами, тем меньше силы взаимодействия между ними и тем ближе- состояние .реального газа к идеальному. И наоборот, чем ближе состояние газа /к области жидкости, тем силы взаимодействия больше и тем значительнее его отклонение от свойств идеального, газа. Таким образом, при изучении свойств реальных газов необходимо учитывать силы взаимодействия между молекулами и объем самих молекул.
В первом приближении Ван-дер-Ваальс ввел в своем уравнении две поправки, которые учитывают отклонение реального газа от идеального. •
Рассмотрим первую поправку, зависящую от объема самих молекул. Уравнение Клапейрона можно представить в виде
V = НТ!р.
При увеличении давления объем и будет уменьшаться, и если р-> со, то V ->- 0. Это полностью согласуется с определением идеального газа, в котором молекулы занимают бесконечно малый объем.
2*
35
Если же рассматривать реальный газ, у которого молекулы занимают конечный объем Vмoл, и учитывать объем зазоров между молекулами при их полной упаковке узаз, то свободный объем для движения молекул будет равен и — Ь, где Ь = умол + узаз.
Величина Ь — тот наименьший объем, до которого можно сжать газ.
При этих условиях уравнение Клапейрона принимает другой вид:
V — Ь = ЯТ/р.
Если в полученной зависимости давление р увеличивается, и стремится к оо, то свободный объем V — Ь стремится к нулю или V -> Ь, т. е. при р -> оо объем газа стремится к величине Ь, которая зависит от объема самих молекул. Для каждого газа величина Ь принимает определенное числовое значение.
Поскольку давление идеального газа по уравнению Клапейрона - определяется как
р = Я77»,
а для реального, газа — с учетом величины Ъ: % , . . Р - ЯТЦр — Ь),
то при одинаковой температуре давление в реальных газах будет больше.'
Это объясняется тем, что у реального газа свободный объем будет меньше, чем у идеального газа, а следовательно, будет меньше и длина свободного пробега молекул, что приведет к большему числу соударений молекул реального газа о стенки, т е." к повышению давления.
Вторая поправка, вводимая в уравнение состояния, учитывает влияние сил взаимодействия между молекулами.
В идеальном газе молекулы практически .свободны в своем движении и удары о стенку сосуда ничем не ограничены, так как сил взаимодействия между молекулами не имеется. -
В реальном газе при наличии сил взаимодействия между молекулами сила ударов о стенку сосуда будет меньше, вследствие того что все молекулы у стенки сосуда притягиваются соседними молекулами внутрь сосуда. Следовательно, и давление, оказываемое реальным газом по сравнению с идельным, будет меньше на величину Ар, которая представляет поправку на' давление, учитывающую силы взаимодействия между молекулами. Эта поправка Ар прямо пропорциональна как числу притягиваемых, так и числу притягивающих молекул, или прямо пропорциональна квадрату плотности газа, или обратно пропорциональна квадрату его удельного объема:
Ар = ар2 = о/у2, . •
*
При этом считается, что молекулы
реального газа представляют собой
иедеформируемые шары.
Вводя вторую поправку, получаем
ЯТ Л ЯГ га\ р — Др или р = . 1
* , V—Ь и — Ь V2
Отсюда уравнение Ван-дер-Ваальса принимает вид, :> (р + а№){о — Ь) = #7\ . (4-1)
• Это уравнение было опубликовано Ван-дер-Ваальсом в 1873 г.
Величину а!ь% называют внутренним давлением: для жидкостей оно принимает очень большое значение (для воды при температуре 293° К , *|г/о « 10800 бар): для газов внутреннее давление сравнительно неве-\ли,ко-И зависит от давления и температуры газа.
Уравнение Ван-дер-Ваальса качественно верно отображает поведение реальных веществ в жидком или газообразном состоянии. Для ^Двухфазных состояний оно неприменимо.
X Для 1 кмоль газа уравнение Ван-дер-Ваальса запишется так: •
Ц-' (Р + я>Ж - М = 8314,2 Т.
![]()
др \ Я'Т . 2а
дv /т {V — Ь)2
дТ )р а .. 2аЬ
р—
V
дТ \ V-
др Я § 4-3. Анализ уравнения Ван-дер-Ваальса
Зьг* Если в уравнении Ван-дер-Ваальса
(Р + аЬг)(ь — Ь) = ИТ. '
"раскрыть скобки и расположить полученные величины по убывающим ^степеням V, то получим уравнение третьей степени относительно удельного объема газа.
ри3 — (Ьр + ЯГ) V2 + аь — аЬ = 0.
" Как известно из математики, такое уравнение при заданных значениях р и Т должно иметь три корня. При этом возможны три случая: .1) вчсе три корня различны и действительны; 2) все три корня действительны и равны между собой и 3) один корень действительный и два . мнимых (комплексных). В последнем случае, поскольку мнимые корни %е имеют физического смысла, реальное значение имеет только один действительный корень.
-Если
на ру-диаграмме построить изотермы,
соответствующие уравнению Ван-дер-Ваальса,
то они будут иметь вид кривых, изобра
женных на рис. 4-3. Израссмотрения этих кривых видно, что при срав- нительно низких температурах они имеют в средней части волнообраз- ный хара^ер с максимумом и минимумом. При этом чем выше темпе- ратура, тем короче становится волнообразная часть изотермы. Прямая АВ, пересекающая такого типа'изотерму, дает три действительных зна- чения удельного объема в точках А, Я и В, т. е. эти изотермы соответ- ствуют первому случаю решения уравнения Ваи-дер-Ваальса (три различных действительных корня). Наибольший корень, равный удель- ному объему в точке В, относится к парообразному (газообразному) состоянию, а наименьший (в точке А) — к состоянию жидкости. По- скольку, как указывалось ранее, уравнение Ван-дер-Ваальса в прин- ципе не может описывать двухфазных состояний, оно указывает (в виде вол- нообразной кривой) на непрерывный переход из жидкого состояния в па- рообразное при данной температуре. В действительности,, как показывают многочисленные эксперименты, пере- ход из жидкого состояния в парооб- разное всегда происходит через двух- фазные состояния вещества, представ- ляющие смесь жидкости и пара. При _ этом при данной температуре процесс
Г 7? ~ ^^^ч»'' 'к перехода жидкости в пар происхо- жу дит также и при неизменном дав- лении.
Практически для особо чистых веществ возможно осуществление участков волнообразной кривой АО_ и ОВ. В первом случае имеют место неустойчивые состояния перегретой жидкости, а во втором — переохлажденного пара. Участок же кривой <2#0 вообще осуществлен быть не может, так как это противоречит условию термодинамической устойчивости, согласно которому для однородного вещества частная производная (др/дю)т не может быть больше нуля.
Положение действительной линии процесса перехода из жидкости в пар изображено на рис. 4-3 прямой линией А В. При этом точка А соответствует состоянию кипящей жидкости, а точка В — состоянию так называемого сухого насыщенного пара, т. е. состоянию, в котором закончился процесс перехода из жидкости в пар. Ветвь изотермы, расположенная правее точки В, соответствует состоянию перегретого пара при данной температуре.
При определенной температуре, называемой критической, изотерма уравнения Ван-дер-Ваальса не будет иметь волнообразного участка. На этой изотерме есть точка перегиба, касательная к которой должна* быть горизонтальной. Это соответствует второму случаю решения уравнения"Ван-дер-Ваальса, когда все три корня действительны и р,ав-ны между собой (рис. 4-3, точка К}'
При температурах выше критической (Т > Тк) изотермы будут
./иметь монотонно спадающий характер, приближаясь по мере увеличения температуры к кривым вида гиперболы. При этих температурах
.(имеет место третий случай решения уравнения Ван-дер-Ваальса, когда ,0дин корень действительный, а два мнимых. Если соединить все точ-%я А, Аъ Л2 и т. д., то получится кривая, на которой жидкость находится в состоянии кипения. Кривую АК называют пограничной кривой жидкости. Соответственно кривая ВК, называемая пограничной кривой пара, представляет собой совокупность состояний сухого насыщенного пара. Таким образом, для реального вещества ро-диаграмму можно ^разбить на три характерные области: 1) область жидкого состояния, расположенную левее пограничной кривой жидкости; 2) область двухфазных состояний (влажного пара), расположенную между пограничными кривыми жидкости и пара, и 3) область перегретого пара,
Чрасположенную правее пограничной кривой пара и выше критической точки. Условно область жидкости ограничивают сверху линией КМ, представляющей собой критическую изобару (линию постоянного дав-
• тения, равного критическому).
В 1869 г. Эндрюс впервые на основании проведенных им экспериментов по изотермному сжатию углекислоты построил ро-диаграмму для реального вещества и показал в ней характерные линии и области. Поэтому часто ри-диаграмму реального вещества называют диаграммой Эндрюса.
Остановимся несколько подробнее на понятии критического состояния вещества.
;; Критическое состояние вещества впервые было открыто Д. И. Менделеевым в 1861 г. Критическую температуру Д. И. Менделеев назвал ■абсолютной температурой кипения, при которой поверхностное натя-гжение в жидкости становится равным нулю, т. е. исчезает различие /между жидкостью и парообразным состоянием вещества (насыщенным „паром).
Д. И. Менделеев дал следующее определение: «Абсолютной температурой кипения я называю такую температуру, при которой частицы .жидкости теряют свое сцепление (поднятие в капиллярной трубке рав-Йно нулю, скрытое тепло равно нулю) и при которой жидкость, несмотря ; "ни на какое давление и объем, вся превращается в пар». Многочисленные опыты с реальными газами полностью подтвердили существование критической точки, в которой исчезает различие между газообразной ,и жидкой фазами.
Из анализа уравнения Ван-дер-Ваальса применительно к критическому состоянию можно получить выражение критических параметров через константы уравнения а и Ь или же определить константы а и Ь при известных критических параметрах.
Учитывая, что уравнение Ван-дер-Ваальса только качественно верно описывает поведение реальных веществ, константы а и Ь обычно вычисляют на основании экспериментальных данных.
Исходным положением для получения зависимости между критическими параметрами и константами уравнения Ван-дер-Ваальса является то, что в критической точке изотерма имеет перегиб и касательная в точке перегиба горизонтальна. Из этих условий вытекает, что
первая частная производная от давления по объему при постоянной температуре (др/ди)тк, а также вторая производная (д1р'1дуг)тк в этой точке должны быть равны нулю. Тогда
(др/ди)тк = —ЯТК У(1»к - 6)а + 2а/ук = 0; (а) (д*р/ди2)Тк = 2/?7У(о* — б)3 — 6й/у4к = 0, (б)
где Г/с и у/с — температура и удельный объем в критической точке. Из уравнения (а) получаем
№/(у/с - Ь? = 2а/и\, (в)
а из уравнения (б) следует
. 2ЯТк/(ик - Ь)3 = 6а/ик. (г)
После преобразований уравнений (в) и (г) имеем
ик = 36. - (4-2)
Подставив значение ик в уравнение (а), получим
Тк = 8а/27(*Ь. (4-3) Из уравнения Ван-дёр-Ваальса следует, что
р = я 7У(у« - 6) — й/ук. Подставляя значение 7\ и у^, находим
рк = я/2762, (4-4)
где Рк — давление в критической точке.
9 г,™ 2' "
'-к
,т*Гкик
= — —
(4-5)
9 ^ 27 /?27>
Рк
Уравнение Ван-дер-Ваальса можно представить в приведенных параметрах состояния. Если вместо переменных р, V и Т ввести в уравнение Ван-дер-Ваальса относительные величины
у/у* = ф, р!рк = я и Т1ТК = т,
называемые приведенными объемом, давлением и температурой, и значения а, Ь и я, выраженные через критические параметры, то получим новое уравнение в следующем виде:
(л + 3/ф2)(3ф — 1) = 8т.
Полученное уравнение называется приведенным уравнением. Оно не включает никаких величин, характеризующих данное вещество* поэтому уравнение справедливо для любого вещества, которое подчи-
,^няется уравнению Ван-дердВаальса. Состояния веществ, находящихся при одинаковых л, ц> и т, называются соответственными состояниями.
В критической точке все три. приведенных параметра имеют одинаковое значение, равное единице, и критические состояния всех веществ являются соответственными.
- Если два вещества имеют одинаковыедва параметра из трех приведенных, то и третий параметр у.этих веществ будет иметь одинаковое ■значение и вещества будут находиться в соответственных состояниях. Указанное явление носит название закона соответственных состояний. Этот закон служит для определения свойств вещества, если известны Свойства другого вещества, находящегося с ним в соответственном состоянии. Такое определение свойств вещества называется методом термодинамического подобия.
Из соотношений для критической точки, полученных из уравнения Ван-дер-Ваальса, следует, что
ЯТк/ркрк = 8/3 = 2,67."
 Это
отношение, обозначаемое Кк>
называют
критическим
коэффициентом. Он
для всех термодинамически подобных
веществ, подчиняющихся уравнению
Ван-дер-Ваальса, должен иметь постоянное
значение, но опытные данные показывают,
что значения Кк
Для
различных реальных газов весьма
отличаются от постоянной величины
(табл.4-1). Это лишний раз подтверждает,
что уравнение Ван-дер-Ваальса правильно
описывает только качественные особенности
свойств газообразных реальных тел..
Это
отношение, обозначаемое Кк>
называют
критическим
коэффициентом. Он
для всех термодинамически подобных
веществ, подчиняющихся уравнению
Ван-дер-Ваальса, должен иметь постоянное
значение, но опытные данные показывают,
что значения Кк
Для
различных реальных газов весьма
отличаются от постоянной величины
(табл.4-1). Это лишний раз подтверждает,
что уравнение Ван-дер-Ваальса правильно
описывает только качественные особенности
свойств газообразных реальных тел..
До открытия критического состояния тела-газы пытались превращать в жидкость только одним увеличением давления, но так как опыты :п.роводйлись при комнатной температуре, то эти попытки успеха не имели.
Опыты показали, .что для превращения газа в жидкость необходимо сначала газ охладить до температуры ниже критической и только после этого сжатием по изотерме можно любой газ превратить в жидкость. *
§ 4-4. Уравнение состояния для реальных газов М. П. Вукаловича и И. И. Новикова
Уравнение Ван-дер-Ваальса при больших плотностях газа дает значительные ошибки, вызываемые тем, что при его выводе не учитывались некоторые добавочные физические 'явления, и прежде всего так называемая силовая, ассоциация и диссоциация молекул.
Кроме того, опытами было доказано, что коэффициенты а и Ь, входящие в уравнение Ван-дер -Ваальса, не могут быть постоянными величинами, а должны зависеть от температуры и давления, причем зависимость эта очень сложная.
Однако попытки многих ученых скорректировать уравнение Ван-дер-Ваальса введением дополнительных зависимостей, учитывающих переменность а и Ь, не позволили существенно расширить область его применения.
Советские ученые М. П. Вукалович и И. И. Новиков в 1939 г. предложили новое универсальное уравнение состояния реальных газов, качественно отличное от уравнения Ван-дер-Ваальса. При выводе своего уравнения авторы учитывали указанное выше явление силовой ассоциации молекул под влиянием межмолекулярных сил взаимодействия.
При явлении ассоциации происходит объединение отдельных молекул в группы, состоящие из двух, трех, четырех и более одиночных молекул. Отдельные молекулы, входящие в группы сложных частиц, сохраняя свои индивидуальные свойства, не реализуют полностью всех степеней свободы. Следовательно, под ассоциацией молекул понимается простое механическое объединение, двух, трех, четырех и более молекул в одну сложную частицу, которая в некоторых отношениях ведет себя как самостоятельная газовая частица. Совокупность однородных газовых частиц, образующихся в результате ассоциации молекул, можно рассматривать как обычный газ. А любой реальный газ рассматривать как смесь нескольких газов, частицами которых являются одиночные, двойные, тройные.и т. д. группы молекул. Эти газы находятся в постоянном взаимодействии друг с другом, и каждый из них достаточно точно следует уравнению Ван-дер-Ваальса. Применяя к подобным газам закон действующих масс и считая, что ассоциация приводит к созданию групп из двух, трех и четырех молекул, М. П. Вукалович и И.И. Новиков получили уравнение состояния, которое было взято за основу при создании первых отечественных таблиц воды и водяного пара, выпущенных М. П. Вукаловичем в 1940 г. В дальнейшем это уравнение было существенно уточнено и. пределы его применения значительно расширены. В наиболее простой форме, когда учитываются лишь
(р+^)(»-*>-*^1-
С
3-f 2т
vT 2
где а и b — постоянные уравнения Ван-дер-Ваальса; Сит — постоянные, определяемые на основании опытных данных,
§ 4-5. Частные производные параметров состояния.' Термические коэффициенты
Если известно уравнение состояния, то каждый параметр состояния может быть выражен как функция двух других параметров, т, е.
р = h (v, Т); т = и (Р> о): о Чв(р, Т). Полные дифференциалы этих величин будут:
dp = (dp/dv)T dv + (dp/dT)v dT,)
dT = (dT/dp)v dp + (dT/dv)pdv, (4-7)
dv = (dv/dp)Tdp + (dv/dT)pdT.}
Частные производные при дифференциалах dp, dT и dv являются попарно величинами взаимно обратными и согласно правилам дифференциального исчисления между ними имеется следующая зависимость:
(dv/dp)T (dp/dv)T = 1; (dT/dv)p(dv/dT)p = 1; (др/дТ),(дТ/др)и = 1.
Следовательно, независимыми частными производными будут три из них. В качестве этих независимых производных выбирают следующие:
(dp/dT)v, (dv/dp)T и (dvldT)p,
Эти частные производные входят в уравнение термических коэффициентов— сжатия, расширения и тепловой упругости, которые могут быть определены опытным путем. .
Указанные частные производные не являются независимыми. Каждая из них может быть выражена как функция двух остальных.
Если уравнение
dp = (dp/dv)Tdv + (dp/dT)vdT рассматривать при р = const, то
(dp/dv)Tdvp + (др/дТ)ЛТР = О,
или
(dp/dv)T + (dpldT)b{dTldv)p = О,
откуда
(dp/dv)r (dv/dT)P = -(др/дТ)0 = -щ^,
или
(др/до) т(дЫдТ)р(дТ/др)„ = . Из последнего уравнения находим
■ (д01дТ)р(дТ/др)г;=
или
(ди1др)т = -^1^. (4-8)
Соотношение (4-8) дает возможность установить связь между изотермическим коэффициентом сжатия р(, термическим коэффициентом расширения тела ар и термическим коэффициентом давления у(. "Эти величины, измеряемые достаточно точно в эксперименте, имеют важное значение для характеристики свойств реальных тел.
Отношение частной производной (дУ~/др)т к объему V характеризует скорость изменения объема с увеличением давления при постоянной температуре. Отношение называют изотермическим коэффициентом сжатия тела
рт = — Ш (дУ/др)т, (4-9)
Знак минус в правой части равенства поставлен для того, чтобы рт получился положительной величиной, так как (дУ/др)т всегда отрицательна.
Отношение частной производной (дУ~/дТ)р к объему V характеризует скорость изменения-объема при нагревании, если давление остается постоянным. Это отношение называют коэффициентом термического расширения тела
ар = 1/У (дУ1дТ)р. (4-10)
Отношение частной производной (др!дТ)у к давлению р характеризует интенсивность изменения давления при увеличении температуры, если объем тела остается постоянным.
Это отношение называют коэффициентом тепловой упругости -или термическим коэффициентом давления
у, = Мр (др/дТ)у (4-11)
Подставляя-значение частных -производных в уравнение (4-8), получим
—рг = —ар/утР, откуда термический коэффициент давления равен
Р Рт ' А,
Коэффициенты ар и рт- определяют из эксперимента, Для идеальных газов -
аР = Рг = 1/Т = 1/273,15,
Контрольные вопросы к IV главе
Чем отличаются реальные газы от идеальных?
Что называется коэффициентом сжимаемости?
Уравнение состояния реальных газов с вириальными коэффициентами.
Что положено в основу вывода уравнения Ван-дер-Ваальса?
Какой смысл имеет константа Ь уравнения Ван-дер-Ваальса?
Какая величина называется внутренним давлением газа?
Уравнение Ван-дер-Ваальса для-1 кг газа.
Проведите исследование уравнения Ван-дер-Ваальса.
Объясните значения корней объема при различных состояниях вещества, полученные из уравнения Ван-дер-Ваальса.
Кто впервые доказал существование критической точки?
Как вычисляются константы а и Ь в уравнении Ван-дер-Ваальса через критические параметры?
При каких условиях можно превращать газы в жидкое состоя- ние? . ' "
Уравнение Ван-дер-Ваальса в приведенных параметрах.
Закон соответственных состояний.
15". В чем заключается силовая.ассоциация молекул?
Что положено в основу вывода уравнения состояния М. П. Ву-каловича и И. И/ Новикова?
Что такое коэффициент изотермического сжатия?
Что такое коэффициент термического расширения тела?
Что такое термический коэффициент давления?
Как связаны между собой термические коэффициенты?