

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf118 |
VECTORS 3 |
|
|
BamHI
AMPR |
TETR |
pBR322
BamHI BamHI
Insert
ori
rop
Cut with BamHI and ligate
BamHI
I n s
e
r t
AMPR
Recombinant plasmid
BamHI
TETR
ori
rop
Transform E. coli
Colony containing plasmid
Petri dish containing agar with ampicillin
Replica plate
Colony containing  non-recombinant plasmid
non-recombinant plasmid
Petri dish containing agar with ampicillin
and tetracycline Colony containing recombinant plasmid on ampicillin plate
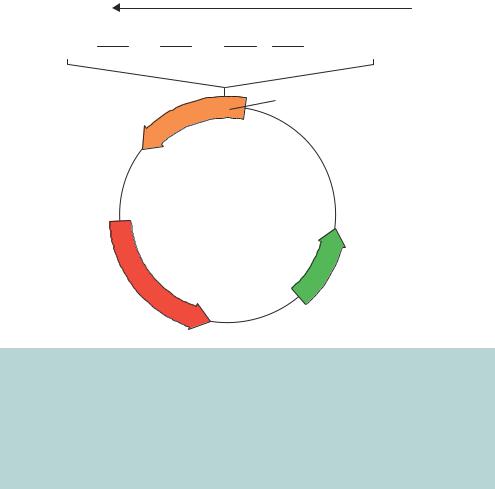
Figure 3.4. Insertional inactivation of antibiotic resistance genes in pBR322. If a DNA fragment is inserted into the BamHI restriction enzyme recognition site within the TETR gene, the resulting recombinant plasmid, when transformed into bacteria, will still give rise to ampicillin resistance, but will be unable to promote bacterial growth on plates containing tetracycline. The TETR gene will be functionally inactivated by the presence of the insert DNA
3.1 PLASMIDS |
119 |
|
|
3.1.2pUC Plasmids
pBR322 was a breakthrough for molecular biology as the first widely used plasmid for molecular cloning, but the double screening procedure required to identify recombinant clones was both time consuming and error prone. In 1982, a new series of plasmids were developed that permitted the identification of the foreign DNA containing cells in a single screening step. These are called the pUC plasmids (Vieira and Messing, 1982). They have three important additional features compared with pBR322 (Figure 3.5).
•High copy number – a mutation within the origin of replication produces 500 –600 copies of the plasmid per cell without the need for chloroamphenicol amplification. The mutation, a G to A change one
lacZ
....L S A H R C |
T S E L |
P D G P |
V S S S N T I M T M |
||||||
HindIII |
PstI |
|
XbaI |
|
SmaI |
|
|
SacI |
|
5'-ccAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCgtaatcatggtcat-3' 3'-ggTTCGAACGTACGGACGTCCAGCTGAGATCTCCTAGGGGCCCATGGCTCGAGCTTAAGcattagtaccagta-5'
SphI HincII BamHI KpnI EcoRI
lacZ′  Multiple cloning site
Multiple cloning site
pUC18 2686 bp
ori
AMPR
Figure 3.5. The pUC plasmids. pUC18 is a small plasmid that contains a mutated version of the ColE1 replication origin that promotes high-copy-number DNA replication. The plasmids also contain the ampicillin resistance gene (AMPR) and the lacZ gene, which encodes the first 63 amino acids of lacZ – the α-peptide. Embedded within the coding sequence of lacZ are the recognition sites for a number of restriction enzymes. This multiple cloning site (or polylinker) is used to clone in DNA fragments. The presence of insert DNA will disrupt the function of the lacZ α-peptide and is used for screening. Different pUC plasmids differ in the composition of the multiple cloning site

120 |
VECTORS 3 |
|
|
Blue precipitate
Non-enzymatic
H dimerisation and N
oxidation
|
|
|
H2O |
CH2OH |
|
H |
|
|
|
Br |
OH |
|
OH |
|
|
|
CH2OH |
O |
N |
|
|||
|
|
|
|
||||
|
|
|
|
|
|
||
OH |
O O |
Cl |
|
OH |
+ |
|
|
|
|
|
|
||||
|
OH |
XGal |
|
|
|
|
Br |
|
|
|
OH |
|
OH |
|
|
|
|
(Colourless) |
|
|
Cl |
||
|
OH |
|
Galactose |
|
5-bromo-4- |
||
|
|
|
|
|
chloro-indoxyl |
||
|
|
|
|
|
|
||
|
|
Tetramer-active |
|
|
|
|
|
|
|||
|
|
|
Wild-type protein |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
||||
lacZ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1-1173 |
|
|
|
a-peptide |
|
|
|||||
|
|
|
|
|
|
|
|
pUC plasmid |
|||
|
|
|
|
|
|
|
|
|
|
|
|
lacZ∆M15 |
|
|
|
|
|
|
|
1-63 |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
42-1173 |
|
|
|
|
|
|
|
|
||
|
|
|
w-peptide |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dimer-inactive |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|

3.1 PLASMIDS |
121 |
|
|
nucleotide upstream of the initiation site of RNAI, reduces the level of the RNAI transcript and consequently results in an increase in DNA replication using RNAII as the primer.
•Blue–white screening – screening of this type is a special form of insertional activation that can be used during the primary selection of transformants, rather than requiring a second round of screening. It utilizes the aminoterminal portion of E. coli β-galactosidase (called the α-peptide) encoded by the vector in a form of intermolecular complementation that restores β-galactosidase activity to a defective enzyme (the ω-peptide) encoded by the host. The E. coli enzyme β-galactosidase is a large polypeptide (monomer = 1173 amino acids; 117 kDa) that is the product of the lacZ
gene. The active form of the enzyme is a homo-tetramer (468 kDa). Certain mutations in the 5 region of lacZ prevent subunit association of the resultant protein (ω-peptide) and the monomers lack enzyme activity (Ullmann, 1992). In some such mutants, subunit assembly (and enzyme activity) can be restored by the presence of a small (50or so amino-acid) amino-terminal fragment of the lacZ product (the α-polypeptide) (Juers et al., 2000). Such lacZ mutants are said to be subject to α-complementation. The product of the lacZ M15 allele lacks amino acids 11 – 41 of wild-type β-galactosidase and is subject to α-complementation. Messing and co-workers took advantage of α-complementation in constructing the pUC plasmid series of cloning vectors (Vieira and Messing, 1982). These vectors carry a multiple cloning site (MCS) embedded in the sequence for the α-peptide gene fragment. The MCS does not alter the reading frame of lacZ or destroy the ability of the fragment to α-complement (Figure 3.6). The insertion of other DNA fragments into the MCS will, however, interrupt the coding sequence of the α-peptide, rendering it non-functional. If a chromogenic substrate (XGal, 5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside) and a β-galactosidase
Figure 3.6. α-complementation and XGal staining. XGal (5-bromo-4-chloro-3-indolyl- β-D-galactoside) is cleaved by a functional β-galactosidase enzyme into galactose and a 5-bromo-4-chloro-indoxyl derivative. The indoxyl spontaneously dimerizes and oxidizes to form an insoluble blue dye (5,5 -dibromo-4,4 -dichloro-indigo; not shown). The β- galactosidase enzyme is the product of the lacZ gene and the active form of the enzyme is a tetramer of identical polypeptides. Certain mutants of lacZ (such as lacZ M15) produce versions of the protein that do not include the extreme amino-terminal end of the 1173 amino acid polypeptide. Such derivatives, termed the ω-peptide, are unable to form the active tetramer and are not functional as β-galactosidase enzymes. The ω-peptide can be made functional by co-expressing the lacZ α-peptide (amino acids 1–63) in the same cell. The α-peptide promotes tetramer formation and restores enzymatic function
122 |
VECTORS 3 |
|
|
inducer (IPTG) are included in the plates on which the primary transformants are selected, non-recombinant molecules will catabolize the colourless substrate to give blue colonies, while recombinants will give white colonies.
•A multiple cloning site or polylinker. This is a synthetic piece of DNA that harbours the sequence of several unique restriction enzyme recognition sites. It was inserted within the portion of the vector encoding the β-galactosidase α-peptide in such a way that it does not affect its expression or function. However, inserting a foreign DNA fragment into any one of the polylinker restriction enzyme recognition sites invariably disrupts the activity of the α-peptide. Thus recombinant colonies remain white but non-recombinants turn blue.
The major cloning advantage of pUC plasmids over pBR322 is that foreign DNA fragments can be cloned into a variety of restriction enzyme sites and recombinants rapidly screened. Additionally, α-complementation requires only a small gene to be carried on the plasmid. The DNA encoding the α-peptide in the pUC-series of vectors is less than 400 bp in length. If the entire lacZ open reading frame were needed, over 3500 bp of DNA would need to be maintained on these vectors. In general, the stability of many plasmid vectors decreases as their size increases. This results in limiting the length of DNA that can be cloned into any particular plasmid. For example, the ability of bacterial cells to maintain recombinant pUC plasmids decreases significantly as their size approaches 15 kbp (Table 3.1). Since the α-polypeptide is small, the total size of the vector is minimized, allowing it to carry a correspondingly larger insert.
3.2Selectable Markers
An essential feature of the plasmids we have discussed so far is the ability to accurately select for cells that have taken up the plasmid. Many such selection systems are currently available. The choice of selectable marker usually rests with the type of cell that is being transformed. Some of the markers will only function against prokaryotes, while others have a broader spectrum of action. Some of the commonly used selectable markers are listed below, together with their mechanism of action.
•Ampicillin – binds to and inhibits a number of enzymes in the bacterial membrane that are involved in the synthesis of the gram-negative cell wall. Therefore, proper cell replication cannot occur in the presence of ampicillin. The ampicillin resistance gene (AMPR or bla) codes for the enzyme
3.2 SELECTABLE MARKERS |
123 |
|
|
β-lactamase that is secreted into the periplasmic space of the bacterium, where it catalyzes hydrolysis of the β-lactam ring of the ampicillin. Thus, the gene product of the AMPR gene destroys the antibiotic. Over time the ampicillin in a culture medium or petri dish may be substantially destroyed by β-lactamase. When this occurs, selective pressure to maintain the plasmid is lost and cell populations can arise that lack the plasmid.
•Tetracycline – binds to a protein of the 30S subunit of the ribosome and inhibits ribosomal translocation along the mRNA and thereby interferes with protein translation. The tetracycline resistance gene (TETR) encodes a 399 amino acid outer membrane associated protein of gram-negative cells that prevents the antibiotic from entering the cell. Thus, the drug resistance gene does not destroy the antibiotic. Selective pressure will be maintained throughout the cell culture process to keep the plasmid containing the drug resistant gene.
•Chloramphenicol – binds to the ribosomal 50S subunit and inhibits protein synthesis. The chloramphenicol resistance gene (CMR) codes for chloroamphenicol acetyltransferase (CAT). The CAT protein is a tetrameric cytosolic protein that, in the presence of acetyl coenzyme A, catalyzes the formation of hydroxyl acetoxy derivatives of chloramphenicol that are unable to bind to the ribosome. As with ampicillin, the CMR gene product destroys the antibiotic.
•Kanamycin and neomycin – bind to ribosomal components and inhibit protein synthesis. The KANR gene codes for a protein that is secreted into the periplasmic space and interferes with the transport of these antibiotics into the cell. Like tetracycline resistance, the KANR gene does not destroy the antibiotic.
•Bleomycin and zeocin – glycopeptide antibiotics that bind to DNA and inhibit DNA and RNA synthesis. They are active against most bacteria (including E. coli), eukaryotic microorganisms (e.g. yeast), plant cells and animal cells. The Sh ble gene from the bacterium Streptoalloteichus hindustanus encodes a small protein that confers resistance to zeocin by binding to the antibiotic (Gatignol, Durand and Tiraby, 1988).
•Hygromycin B – inhibits translation by interfering with ribosome translocation. The antibiotic is active against both prokaryotes and eukaryotes. The resistance gene (HYGR, encoding hygromycin-B-phosphotransferase) inactivates the antibiotic by phosphorylation.
124 |
VECTORS 3 |
|
|
Plasmid based vectors are extremely widely used and have been adapted to serve a variety of functions. Many of these will be discussed in later chapters of this book, but here I will list a number of examples to give the reader a flavour of the diversity of plasmid use.
• General cloning – most DNA manipulation performed in the laboratory is carried out in plasmids. The ease of both use and storage of plasmid DNA molecules makes them a popular choice for most recombinant DNA experiments.
•Shuttle vectors – these plasmids contain not only the origin of replication and selectable marker for E. coli, but also functionally similar sequences for maintenance in other hosts. For example, plasmids for the cloning and expression of genes in the yeast Saccharomyces cerevisiae contain both replication origins and selectable markers for both E. coli and yeast. Most DNA manipulations will be performed using E. coli as a host, prior to transformation of the final DNA construct into yeast.
•RNA production – many plasmids have been designed so that the foreign DNA fragments cloned into then can be transcribed into RNA. Such plasmids contain the promoter sites for an RNA polymerase, e.g. those from the bacteriophages T3, T7 or SP6, such that RNA can be made in vitro using the purified RNA polymerase and the plasmid DNA. The RNA made by this method is often used as probes for hybridization in Northern blotting.
•Protein production – many plasmids contain promoter sequences to express the foreign genes that they contain. Often the expression is performed
in E. coli, but, using the appropriate promoters, protein expression can be driven in almost any organism. High-level protein production could be driven from a strong promoter, while low-level production would be driven from weaker promoters. Levels of protein production may also be modulated by altering the copy number of the plasmid.
Given the wide range of plasmids that are available to researchers today, systems have been developed to move DNA fragments between a variety of plasmids with differing functions. For example, if you have cloned a gene, you might want to express the gene product at high levels in E. coli, while also expressing the protein in mammalian tissue culture cells and producing a tagged version of the protein to which monoclonal antibodies are available. Systems have therefore been devised for the shuttling of DNA fragments between
3.2 SELECTABLE MARKERS |
125 |
|
|
AMPR
ori
loxP |
T |
|
|
|
a |
|
r |
|
g |
|
e |
|
t |
Donor |
g |
e |
|
plasmid |
n |
e |
|
loxP |
|
Mammalian
promoter AMPR
loxP
Acceptor plasmid
ori
Mix and add Cre recombinase
|
|
|
|
e |
|
|
|
|
n |
|
|
|
|
e |
|
|
e |
t |
g |
|
g |
|
||
r |
|
|
||
|
|
|
||
|
|
|
|
|
a |
|
|
|
|
T |
|
|
|
|
 loxP
loxP
ori
Mammalian promoter
loxP
Acceptor
plasmid
AMPR
loxP: 5'-ATAACTTCGTATAGCATACATTATACGAAGTTAT-3'
3'-TATTGAAGCATATCGTATGTAATATGCTTCAATA-5'
13 bp repeat |
8 bp |
13 bp repeat |
|
spacer |
|
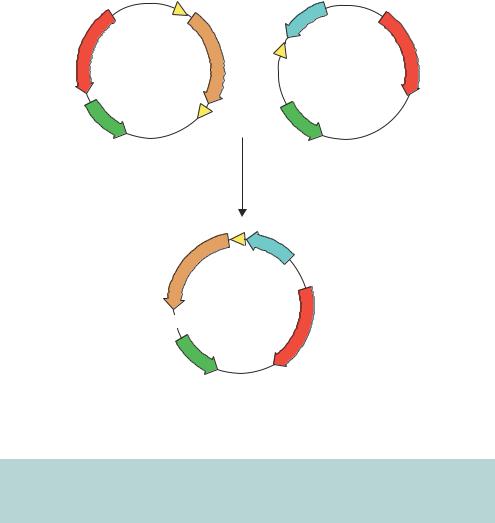
Figure 3.7. Gene shuffling between plasmids using recombination. Genes are transferred from the donor plasmid to the acceptor plasmid at the loxP sites using the Cre recombinase. See the text for details
vectors without having to use restriction enzymes. One such system is outlined in Figure 3.7. Here, the DNA fragment encoding the target gene is transferred from one plasmid to another by the action of the Cre recombinase. Cre is a 38 kDa recombinase protein from the bacteriophage P1 (Sternberg et al., 1981). It mediates recombination between or within DNA sequences at specific locations called loxP sites (Abremski and Hoess, 1984). These sites consist of two 13 bp inverted repeats separated by an 8 bp spacer region. The 8 bp spacer region in the loxP site has a defined orientation that forces recombination to occur in a precise direction and orientation. Donor plasmids contain two loxP sites, which flank the target gene. Acceptor plasmid contain a single loxP site and elements to which the target gene will become fused. The target gene, once
126 |
VECTORS 3 |
|
|
transferred, will become linked to the specific expression elements for which the acceptor vector has been designed. Furthermore, if the coding sequence for the gene of interest is in frame with the upstream loxP site in the donor vector, it will automatically be in frame with all peptides designed in the acceptor vector. An alternative donor and acceptor plasmid system is based upon site-specific recombination reactions mediated by phage λ (Karimi, Inze and Depicker, 2002). In this case, DNA fragments flanked by recombination sites (att) can be transferred into vectors containing compatible recombination sites (att × attP or attL × attR) in a reaction mediated by the λ recombination proteins.
The versatility of plasmids has lead to their widespread acceptance as the vectors of choice for many gene manipulation experiments. Plasmids do, however, suffer from a number of significant shortcomings. First, the efficiency at which the plasmid is transferred to a bacterial cell is very low. Plasmid DNA molecules must be transformed into competent bacterial cells (see Chapter 2), but this process is inefficient. At best, E. coli cells can be made competent to a level such that 1 × 109 transformed cells can be generated per microgram of plasmid DNA. For a typical plasmid, 1 µg represents about 1.5 × 1011 molecules. This means that the cells are taking up less than 1 per cent of the available DNA molecules. Second, the capacity of plasmids to carry large fragments of foreign DNA is limited (Table 3.1). Most plasmids become unstable if their overall size exceeds about 15 kbp. Plasmids larger than this tend to undergo recombination events, which can result in the reordering or elimination of DNA from them. Other types of vector have thus been developed to overcome these difficulties.
3.3λ Vectors
In the early 1950s, Andre´ Lwoff described an astonishing property of Escherichia coli. When he irradiated certain strains of E. coli cells with a moderate dose of ultraviolet light, the bacteria stopped growing and, after about 90 min, the bacteria lysed and released many viral particles, called λ, into the culture media (Figure 3.8). The viruses, more commonly called bacteriophages or simply phages, are able to infect other E. coli cells that had not previously been infected by λ phages. Not all bacterial cells underwent this lytic phase when irradiated in this way. Most E. coli cells are relatively unaffected by such small ultraviolet doses. However, bacteria that had previously been exposed to phage λ, but had not undergone lysis, showed this remarkable property.
Upon infection by phage λ, E. coli cells will undergo one of two fates. Either cell lysis proceeds and newly synthesized phage particles will be released into the surrounding medium, or, alternatively, the phage can switch into a
3.3 λ VECTORS |
127 |
|
|
Head |
|
Viral genome |
|
|
|
Tail
100 nm
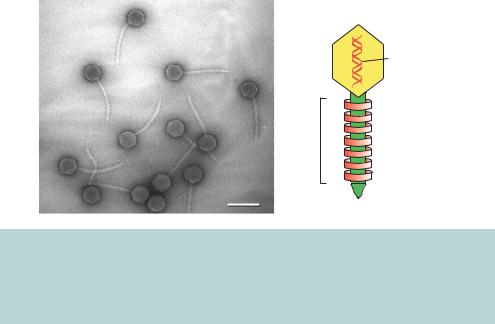
Figure 3.8. The structure of bacteriophage λ. An electron micrograph of λ phages that have been released upon bacterial lysis, and a diagrammatic representation of the overall structure of λ. The EM image is courtesy of Professor Ross Inman (University of Wisconsin) and is reproduced with permission. Other excellent EM images of λ can be found on Professor Inman’s web site (http://www.biochem.wisc.edu/inman/empics/)
dormant lifestyle in which the phage DNA becomes integrated into the E. coli chromosome – in this case a lysogenic lifestyle is adopted. The life cycle of λ phage is shown diagrammatically in Figure 3.9. It is the lysogenic bacteria that show rapid lysis upon ultraviolet radiation. In the laboratory, λ phage growth and replication is monitored on petri dishes (Figure 3.10). The phage is mixed with E. coli cells is a soft agar solution called top agar. The mixture is poured onto the surface of a nutrient agar plate and incubated to allow bacterial growth. λ growth will cause the death (lysis) of the E. coli cells surrounding the site of initial infection. Such sites are observed on the plates as somewhat turbid plaques in the bacterial lawn. λ DNA can be purified from the phage particles contained with these plaques.
The genetics and molecular biology of bacteriophage λ have been extensively studied – for further information on λ, readers are directed to the excellent text by Mark Ptashne (Ptashne, 1992). The DNA contained within the phage is a linear double-stranded molecule 48 502 bp in length. The extreme 5 - and 3 -ends of the λ genome have 12 bases that are single-stranded – called the cohesive or cos ends. These sequences are complementary and can anneal with each other to form a circular double-stranded DNA molecule (Figure 3.11). Functionally related genes of λ are generally clustered together on the λ genome, except for the two positive regulatory genes N and Q. Genes on the left-hand side of the conventional genome map code for head and tail proteins of the phage particle. These are followed by genes whose protein products