

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf208 GENE IDENTIFICATION 6
clinically for heart attack sufferers to prevent further blood clotting (Madhani, Movsowitz and Kotler, 1993). In the early 1980s human tissue-type plasminogen activator (t-PA) was purified biochemically from melanoma cells and digested with the protease trypsin. Some of the resulting protein fragments were subjected to amino acid sequencing and one of the peptides produced in this way is shown in Figure 6.2. Using the genetic code (see the Appendix), all possible DNA sequences that could encode this peptide were determined (Figure 6.2). Of the 15 bases within the five codons, only four of them contained potential alternate sequences. Therefore, Pennica et al. constructed a degenerate 14-nucleotide antisense DNA probe, which contained a mixture of eight different sequences (Pennica et al., 1983). They used this probe to screen a plasmid cDNA library of 4600 clones prepared from the melanoma cell line. Of these, 12 were scored as positive in the hybridization screen and, after DNA sequencing, one of these was found to contain the DNA that could have encoded the peptide. This process led to the isolation of the full-length cDNA encoding the t-PA protein.
The DNA probe used in a hybridization experiment must be homologous to the sequence it is to detect. There are examples in the literature where the gene
Amino acid sequence:
-Trp-Glu-Tyr-Cys-Asp-
Possible DNA sequence:
5′-TGG-GAA-TAT-TGT-GAT-3′
G C C C
Probe:
3′-ACC-CTT-ATA-ACA-CT-5′
C G G
Oligonucleotides in probe:
3′-ACC-CTT-ATA-ACA-CT-5′ 3′-ACC-CTC-ATA-ACA-CT-5′
3′-ACC-CTC-ATG-ACA-CT-5′
3′-ACC-CTC-ATA-ACG-CT-5′ 3′-ACC-CTC-ATG-ACG-CT-5′ 3′-ACC-CTT-ATG-ACA-CT-5′ 3′-ACC-CTT-ATA-ACG-CT-5′ 3′-ACC-CTT-ATG-ACG-CT-5′
Figure 6.2. Probe design for the isolation of the human tissue-type plasminogen activator (Pennica et al., 1983). The sequence of five amino acids of the protein is shown. This sequence (representing amino acids 253–257 of the 527-amino-acid protein), was generated following digestion of the isolated full-length protein with trypsin. Based on the genetic code, a degenerate probe was designed that would bind to all possible DNA sequences that could encode this peptide. The probe sequence outlined in yellow represents that which is precisely complementary to the isolated gene
6.1 SCREENING BY NUCLEIC ACID HYBRIDIZATION |
209 |
|
|
isolated from one organism has been used as a hybridization probe to detect a homologous gene in a DNA library generated from a different organism. For example, some of the histone genes encoded by the sea urchin have been used to isolate the homologous histone genes from a frog library (Old et al., 1982). As we have seen above, chemically synthesized degenerate oligonucleotide probes, in the range of approximately 14 –20 nucleotides in length, can be used to detect DNA sequences encoding particular proteins. In the case of t-PA above, the peptide sequence generated could only be coded by one of a few DNA sequences – i.e. a sequence containing few degeneracies. Other amino acids are encoded by up to six different triplet codons. For example, the amino acid leucine is coded for by the following triplets: CTA, CTC, CTG, CTT, TTA and TTG. A larger number of degeneracies in a probe sequence will result in the binding of the probe to sequences that do not encode the intended target gene, whereas a highly specific probe will bind to relatively few gene sequences. Consequently, peptide sequences containing the amino acids methionine and tryptophan, which are each encoded by a single triplet, are particularly important for designing such probes. As we have previously seen with Southern blotting (Chapter 2), the stringency of washing the probe from the membrane can be used to adjust the number of positive interactions that occur.
Hybridization probes are usually radioactively labelled to aid their easy detection when bound to the membrane. Chemically synthesized oligonucleotide probes can be treated with the enzyme polynucleotide kinase in the presence of γ -32P-ATP so that the radioactive phosphate group from the ATP molecule is transferred onto the 5 -end of the oligonucleotide. Other, non-radioactive alternatives have been developed, e.g. labelling with digoxigenin (McCreery, 1997), and are useful for certain experimental procedures, but the sensitivity and detection power of radioactivity has been difficult to surpass.
Colony screening, as described above, can be used to screen plasmid or cosmid based libraries. However, with only slight modifications it can also be used to screen λ phage libraries (Benton and Davis, 1977). Indeed, the screening of λ plaques is considered more desirable.
•Less DNA from the bacterial host will be transferred to the nitrocellulose membrane when lifting plaques rather than bacterial colonies. This results in a ‘cleaner’ background (less background probe hybridization) for λ plaque screening.
•Plaques can be lifted several times, so multiple screens can be performed from the same plate.
•Screening can be performed at very high density by screening small plaques. High-density screening has the advantage that a large number
210 GENE IDENTIFICATION 6
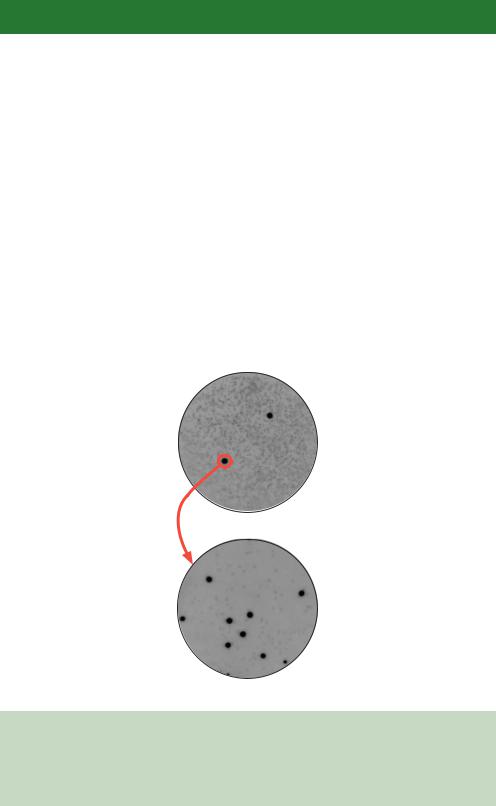
of recombinant clones can be screened for the presence of sequences homologous to the probe in a single experiment. Screening in this way, however, means it is unlikely that a single pure recombinant clone will be isolated from one round of screening. As shown in Figure 6.3, screening an agar plate containing 50 000 or more individual plaques with a probe may generate one, or two, spots in the X-ray film corresponding to positions where the probe has bound. In this case, an area of agar corresponding to the site of the spot on the original agar plate was removed and the multiple plaques contained within it were re-plated at a lower density. The screen was then repeated to generate a secondary hybridization pattern that was enriched for the plaque showing as positive in the screen. Two or three rounds of screening are often required before a pure plaque can be isolated.
A logical extension of hybridization screening is, rather than screening a library with a single oligonucleotide to search for homologous sequences, screening by PCR using two primers to amplify homologous portions of genes. The major
First screen
Second screen
Figure 6.3. Multiple rounds of screening to isolate a single pure recombinant. In a hybridization screen of a λ phage library, positives from the first round of, high-density, screening are re-plated and re-screened at lower density. This allows the isolation of single, pure recombinant plaques. Images courtesy of Michael Bromley and Jayne Brookman, F2G Ltd
6.2 IMMUNOSCREENING |
211 |
|
|
advantage of this approach is speed. The library need not be plated prior to screening since the PCR reaction will occur using naked DNA as a template. Degenerate primers can be used to amplify portions of homologous genes from the library (Takumi, 1997). The isolated PCR products usually represent only a small region of the gene. However, this isolated fragment can be used as a highly specific probe in a traditional hybridization screen, or as a starting point to amplify the 5 - and 3 -ends of the gene using various PCR methods (Frohman et al., 1988).
6.2Immunoscreening
If a DNA fragment library is cloned into an expression vector (see Chapter 3), the gene products encoded within the foreign DNA may be produced within the host cell. Even if the protein is not fully functional, the sequence of the expressed peptide is likely to be unique within the host cell. Therefore, mechanisms to identify these unique polypeptide sequences can be used to screen the library in order to identify particular clones. The detection of polypeptide sequences is usually performed using antibodies. Antibodies are relatively straightforward to produce if a purified, or even partially purified, protein is available. The gene encoding this protein can then be identified using the antibody in screening procedures outlined below. Screening of this type does not rely upon any particular function of the expressed foreign protein, but does require a specific antibody to that protein to be available.
Antibodies are raised when a foreign protein or peptide is injected into an animal. Often, the animal used to raise antibodies for use in the laboratory is the rabbit or mouse, but sheep, goats, pigs and horses have all been used to generate larger amount of antibody (Harlow and Lane, 1999). The presence of the foreign protein (antigen) is detected in the animal by surface receptors on B and T lymphocyte cells. Each B cell has many thousands of different receptors on its surface that are able to bind to particular antigens. The binding of the antigen to an individual receptor results, via a complex pathway, in the descendants of that B cell secreting vast numbers of the soluble form of that particular receptor. These are the antibodies.
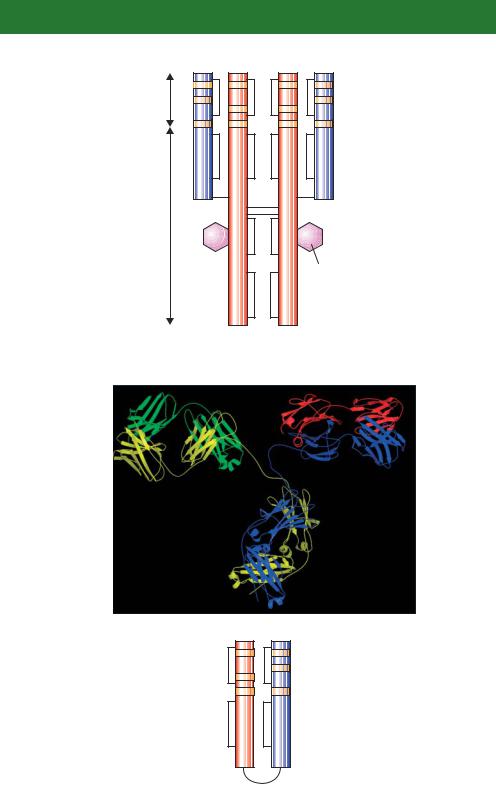
Antibodies are glycoproteins composed of subunits containing two identical light chains (L chains), each containing about 200 amino acids, and two identical heavy chains (H chains), containing about 400 amino acids each (Davies, Padlan and Sheriff, 1990). The amino-terminal 100 or so amino acids of both the H and L chains vary greatly from antibody to antibody – these are termed the variable (V) regions. The amino acid sequence variability in the V regions is especially pronounced at three hypervariable sites (Figure 6.4). The tertiary structure of the antibody brings the three hypervariable regions of both the

6.2 IMMUNOSCREENING |
213 |
|
|
Table 6.1. The five classes of antibodies (Harlow and
Lane, 1999)
Class |
H chain |
L chain |
Subunit structure |
|
|
|
|
IgA |
α |
κ or λ |
(H2L2)2 |
IgD |
δ |
κ or λ |
H2L2 |
IgE |
ε |
κ or λ |
H2L2 |
IgG |
γ |
κ or λ |
H2L2 |
IgM |
µ |
κ or λ |
(H2L2)5 |
L and the H chains together to form an antigen binding site. Only a few different amino acid sequences are found at the carboxy-terminal end of H and L chains – the constant (C) regions. Mammals produce two different kinds of C region for their light chains, kappa (κ ) L chains and lambda (λ) L chains. Additionally, five different kinds of C region for H chains are produced: α (the heavy chain of IgA antibodies), γ (IgG), δ (IgD), ε (IgE) and µ (IgM). Each type of H chain is able to pair with either λ or κ L chains (Table 6.1). Covalent disulphide linkages hold the pairings of H and L chains together.
Antibody molecules are required to perform two functions – they must recognize and bind to an antigen and then trigger the cellular response to that antigen. The V regions are responsible for antigen recognition, while the C regions are responsible for triggering the cellular response. The five different types of heavy chain provide a mechanism for invoking different cellular responses to an antigen (Wysocki and Gefter, 1989).
Most antibodies in use in the laboratory are described as either polyclonal or monoclonal. Polyclonal antibodies are isolated from the serum of an immunized
Figure 6.4. The structure of an antibody. (a) The diagrammatic representation of antibody structure. The heavy chains (red) and the light chains (blue) are connected together via a series of disulphide bridges (black lines). Both the light and heavy chains possess a series of hypervariable regions (orange) at their amino-terminal ends that provide an immense level of antigen binding site diversity. (b) The X-ray crystal structure of a monoclonal antibody. The heavy chains (yellow and blue) and the light chains (green and red) are shown (Harris et al., 1992). (c) A single-chain antibody variable region fragment (scFv) antibody. The variable regions from the heavy and light chains can be engineered to be expressed as a single polypeptide joined by a 15-amino-acid linker (of the sequence (glycine4serine)3), which has sufficient flexibility to allow the two domains to assemble a functional antigen binding site

214 GENE IDENTIFICATION 6
animal and contain many different antibodies that recognize different epitopes of the same antigen. Monoclonal antibodies are produced from isolated, clonal cells and recognize an individual specific epitope within the antigen. Antibodies that bind to proteins can recognize either continuous (i.e. the primary amino acid sequence of the protein) or discontinuous epitopes. A discontinuous epitope is formed by the folding of the protein to generate a surface area antigen that is composed of different segments of the primary structure. The bulk of naturally occurring epitopes are of the discontinuous type, although antibodies for use in the laboratory are often produced using denatured protein to ensure that the produced antibodies recognize continuous epitopes and can thus be used to detect the denatured protein using western blotting (Chapter 2). Although the use of animals in the production of high-affinity antibodies remains widespread, several protocols are available for the selection and production of specific antibody fragments in bacterial cells (Hexham, 1998; Portner-Taliana et al., 2000; Daugherty et al., 1999).
A generalized scheme for the immunoscreening of a DNA library is shown diagrammatically in Figure 6.5. cDNA is cloned into the expression vector λZAP (see Chapter 3) such that the foreign DNA is placed under the control of the bacterial lac promoter. Pooled recombinant λ phages are plated out onto a suitable bacterial host on agar plates. Plates containing the phage library are incubated until small plaques appear. At this point, a nitrocellulose sheet that had previously been soaked in IPTG, the gratuitous inducer of the lac promoter, is placed on top of the plaques. The nitrocellulose sheet is left on top of the agar for four hours to both induce the expression of the polypeptides encoded by the cDNA, and bind the proteins that are produced when the E. coli cells lyse as a consequence of the phage infection. The nitrocellulose sheet is then peeled off the plate and will contain, adsorbed to its surface, the proteins that were expressed in each individual plaque. The sheet is then incubated with a specific antibody to the protein for which the gene is sought. The antibody should only bind to the nitrocellulose sheet in a position where a plaque expressing that protein was located on the original agar plate. The sheet is then washed to remove any unbound antibody and subsequently incubated with a labelled secondary antibody to detect the presence of the bound primary antibody.
Originally, immunoscreening methods involved the use of radio-labelled primary antibodies to detect antibody binding to the nitrocellulose sheet (Broome and Gilbert, 1978). Such methods have, however, been largely superseded using the antibody sandwiches described above. The secondary antibody recognizes the constant region of the primary antibody and is, additionally, conjugated to an easily assayable enzyme. Such enzymes, for example, horseradish peroxidase or alkaline phosphatase (Mierendorf, Percy and Young, 1987; de Wet et al.,


 cDNA
cDNA 


 soaked in IPTG
soaked in IPTG
216 GENE IDENTIFICATION 6
enzyme that catalyses the reversible phosphorolysis of uridine to uracil) from a colon tumour (Liu et al., 1998). Antibodies to the purified protein were raised in rabbits and used to screen a human liver cDNA expression library. This resulted in the isolation of a 1.2 kbp clone that contained the entire open reading frame of the human uridine phosphorylase gene.
A particularly successful combination of membrane based protein function screening, sometimes called south-western screening, has been used extensively to identify genes that encode DNA binding proteins. Screening of this type is performed as outlined in Figure 6.5, except that, rather than incubating the nitrocellulose membrane with an antibody, radiolabelled double-stranded DNA of known sequence is used instead (Singh et al., 1988). Proteins that are able to bind to this sequence, often originating from the promoter of a gene, will trap the DNA on the membrane, and their position can be visualized using X-ray film. This approach requires that DNA binding can occur on the membrane and also requires that the DNA binding activity is contained with a single polypeptide. Nevertheless, it has been used to isolate numerous DNA binding proteins (Vinson et al., 1988).
6.3Screening by Function
The screening of cDNA libraries using antibodies described above relies only on the expression of cDNA encoded polypeptide sequences. It does not require the expression of a fully functional protein. Screening can, however, be performed to identify a specific protein function within the host cell. For this type of screening to be successful, a host cell is required that either lacks a biochemical function that can be selected for, or that is specifically disabled in some function that can be compensated for by a protein produced from an expression library. This functional complementation is particularly useful for identifying genes from one organism that perform the same role as a defective gene in another organism. For example, E. coli cells harbouring a defective copy of the hisB gene, encoding the enzyme imidazole glycerol phosphate dehydratase that is essential for the biosynthesis of the amino acid histidine, are unable to grow on media lacking histidine. If the hisB defective E. coli cells are transformed with an expression library from yeast and plated onto media lacking histidine, the only cells that will be able to grow will be those producing a functional copy of the yeast enzyme – encoded by the HIS3 gene (Ratzkin and Carbon, 1977). The yeast HIS3 and E. coli hisB genes share little DNA sequence similarity (less than 20% overall identity at the amino acid level), but the encoded proteins, although different in amino acid sequence, perform the same enzymatic function.
6.4 SCREENING BY INTERACTION |
217 |
|
|
Functional cloning has been particularly successful in the isolation of higher-eukaryotic genes as functional homologues of genes found in more experimentally amenable lower-eukaryotic cells. For example, many highereukaryotic genes have been isolated by their ability to complement defects in their yeast counterparts. These include
•the genes coding for several human metabolic enzymes, reviewed by Botstein and Fink (1988),
•the Drosophila topoisomerase II gene (Wyckoff and Hsieh, 1988),
•a number of human RNA polymerase II transcription factors (Becker et al., 1991) and
•mouse cell cycle control genes (Martegani et al., 1992).
The major drawback with screening of this sort is that an assayable mutation within the host cell must be available that can be compensated for by the gene expressed from the foreign DNA. For many genes, such assays are simply not available. Additionally, mutations may not be fully compensated for by the foreign gene. For example, the foreign gene may be only partially functional within the host cell. A foreign gene may not be expressed within the host cell, or the produced proteins may not be appropriately post-translationally modified to produce the active form. Also, since library construction usually results in the cloning of single genes into vectors, if two or more different foreign gene products are required to produce the active protein, complementation screening is unlikely to succeed.
Genes may be cloned as a consequence of function if the expressed proteins are able to confer a new phenotype upon the host cell into which they are transformed. For example, cellular oncogenes can be isolated from human DNA libraries based on their ability to stimulate cell proliferation in culture (Brady et al., 1985). This ‘gain of function screening’ may have a limited number of possible uses, but is an extremely powerful way to identify specific genes where it has been applied.
6.4Screening by Interaction
Most proteins do not exist within cells as single entities. Most interact with a range of other proteins that either regulate their function or assemble them into larger functional complexes (Legrain and Selig, 2000). Therefore, once we have cloned a gene that encodes a protein, we might want to ask with which other proteins it interacts.