

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf298 |
GENOME SEQUENCING PROJECTS 9 |
|
|
binding sequences flanking cloning sites so that unknown DNA cloned into them may be sequenced.
•Unlike the Maxam –Gilbert technique, the DNA strand that is being sequenced does not need to be radio-labelled. Labelling is required so that the extended and chain terminated products can be detected after gel electrophoresis. A radio-label (e.g. 32P, 35S, or 33P in the form of an α-modified deoxynucleotide) can be incorporated into the newly extended chain as part of the replication process itself.
•The DNA molecule to be sequenced does not necessarily have to be single stranded. The original Sanger method was used to sequence linear double-stranded restriction digestion products, but was not directly applicable to the sequencing of double-stranded plasmids. This led to the widespread use of M13 vectors to produce single-stranded templates for sequencing (see Chapter 3). The single-stranded DNA produced from M13 generally yielded very clean readable sequence. The Sanger technique was subsequently adapted to allow for the denaturation of plasmid DNA using alkali that was suitable for sequencing (Yie, Wei and Tien, 1993).
Many modifications have been made to the chain-terminating sequencing protocol since its inception, but the basic chain-termination method devised by Sanger has remained the cornerstone of almost all sequencing projects. The Klenow fragment of E. coli DNA polymerase I was originally used as the replicating enzyme, but this was superseded by a modified form of the DNA polymerase from the bacteriophage T7 (also known as Sequenase ), which proved to be a more processive enzyme that allowed more sequence to be read from a single reaction (Griffin and Griffin, 1993). It is essential that an enzyme lacking a 5 –3 exonuclease activity is used for sequencing to ensure the integrity of the newly synthesized DNA fragments. Nowadays, most sequencing is performed using Taq DNA polymerase. The high temperatures at which the thermostable enzyme can replicate DNA ensure that secondary structure is kept to a minimum so that cleaner, more readable sequences can be obtained. The use of Taq DNA polymerase is often combined with thermocycling to amplify a single DNA strand of a duplex in a linear manner from a single primer (Murray, 1989). This eliminates the requirements for separate double-stranded DNA denaturation and primer annealing steps. The method enables sequencing from very small amounts of double-stranded DNA, and also allows direct genomic DNA sequencing from bacterial colonies or phage plaques, thereby bypassing the requirement for cloning entirely (Slatko, 1996).

 3'-
3'-
300 |
GENOME SEQUENCING PROJECTS 9 |
|
|
9.4.2Automated DNA Sequencing
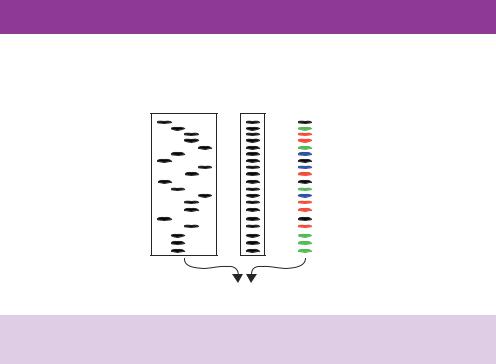
Sequencing using the Sanger technique can lead to clean and unambiguous assignment of about 300 bases per reaction. The method is, however, quite labour intensive. For example, multiple pipetting steps are required to set up each reaction and then the reactions must be loaded onto four lanes of a gel to separate the products. Additionally, the manual reading of sequencing gels (Figure 9.7) can be both time consuming and error prone. To tackle the sequence of the human genome ( 3.2 × 109 bp), more automated and rapid methods of sequence collection were required.
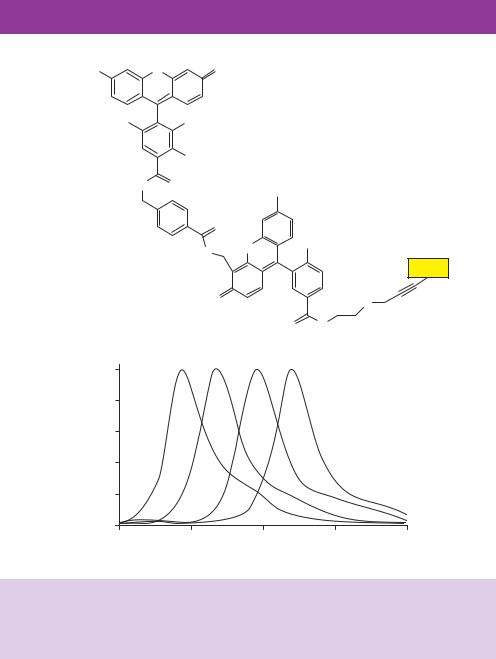
A straightforward way to increase the throughput of DNA sequencing would be to combine the four individual sequencing reactions (each containing a different ddNTP) into a single reaction that could be analysed on a single lane of a gel. This is not possible using radioactivity since each band (Figure 9.7) is distinguishable only by the position in which it runs on the gel. Therefore, combining all four lanes would merely result in a series of bands differing in size by a single base (Figure 9.8). However, if the terminal base of each DNA fragment can be identified specifically then, since each band on the gel is a different size, the DNA sequence can be unambiguously assigned from a single gel lane. A set of dideoxynucleotides has been developed that are labelled with fluorescent dyes precisely for this purpose (Glazer and Mathies, 1997). The dideoxynucleotide can still be incorporated into DNA opposite its complementary base, which again results in the termination of DNA synthesis. The dye structures attached to the dideoxynucleotide contain a fluorescein
donor dye linked to a dichlororhodamine (dRhodamine) acceptor dye via an aminobenzoic acid linker and are called BigDye terminators. An argon ion laser is able to excite the fluorescein donor dye that efficiently transfers the energy to one of the four acceptor dyes, each of which has a distinctive emission spectrum (Figure 9.9). Each dideoxynucleotide is labelled with a different acceptor dye so that DNA fragments ending in a different ddNTP
Figure 9.7. DNA sequencing using dideoxynucleotide chain terminators. DNA replication is initiated from an oligonucleotide primer and four individual sequencing reactions are performed each of which contains all the dNTPs and a single ddNTP (either G, A, T or C, as indicated). DNA replication is terminated when the ddNTP (highlighted in yellow) is incorporated to generate a series of different length DNA molecules that can be separated using a polyacrylamide gel. The sequence of the newly synthesized DNA can be read in a 5 to 3 direction from the bottom of the gel to the top. In the example shown, the primer produces a new ‘bottom’ DNA strand. The sequence of the ‘top’ strand can be obtained from this

304 |
GENOME SEQUENCING PROJECTS 9 |
|
|
sequencing can be performed directly on genomic DNA, this is generally impractical on a large scale due to the large number of different oligonucleotide primers that would need to be synthesized to initiate the sequencing reactions. Genomic DNA fragments are therefore cloned into a vector and each fragment is subsequently sequenced using the automated methods described above. The problem then is how to reconstruct the original genome sequence based on the small fragments that are cloned into individual vectors. Several basic approaches have been used.
•Clone contigs. The simplest way to generate overlapping DNA sequence is to isolate and sequence one clone, from a library, then identify (by hybridization) a second clone, whose insert overlaps with the first. The second clone is then sequenced and the information used to identify a third clone, whose insert overlaps with the second clone, and so on. This is the basis of chromosome walking and was first used to build up large continuous DNA sequences (contigs) from small fragments cloned into vectors. This method is, however, laborious. A single clone has be isolated and sequenced before the next overlapping clone can be sought. Additionally, repetitive sequences within the genome can give rise to incorrect contig assignment.
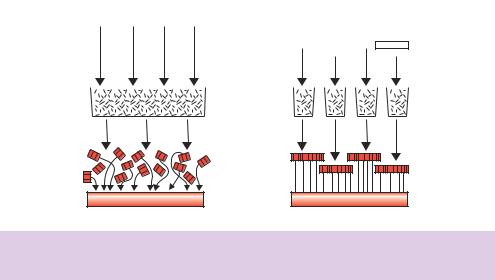
•Whole genome shotgun. The fragments of the genome, which have been randomly generated, are cloned into a vector and each insert is sequenced. The sequence is then examined for overlaps (sequences that occur in more than one clone) and the genome is reconstructed by assembling the overlapping sequences together (Figure 9.11). This approach was first used to sequence the genome of the bacterium Haemophilus influenzae. The entire genome of the organism was randomly fragmented using sonication and then small fragments (in the range of 1.5 –2 kbp) were cloned into a vector (pUC18). The resulting library consisted of approximately 20 000 individual clones. Each of these was then sequenced to generate approximately 12 million base pairs of sequence information (Fleischmann et al., 1995). This corresponds to six times the length of the H. influenzae genome. The sequence obtained from each clone was then assembled into contigs based on the overlaps between the individual clones. Any sequence gaps were filled in subsequently by identifying additional clones (from a different library) that contained sequences close to the gap-point. The main advantage of the shotgun approach is that no prior knowledge of the sequence of the genome is required. The approach is, however, limited by the ability to identify overlapping sequences. Every sequence obtained must be compared with every other sequence in order to identify the overlaps. This can be a time-consuming process and requires large amounts of computational
306 |
GENOME SEQUENCING PROJECTS 9 |
|
|
•determine the DNA sequence of the entire human genome
•store this information in databases
•identify all of the genes in human DNA
•improve tools for data analysis.
The project was planned to last for 15 years, but technological advances have accelerated the expected completion date to 2003. On 26 June 2000, in a joint press conference, President Bill Clinton (USA) and Prime Minister Tony Blair (UK) announced that the first rough draft of the human genome sequence had been completed. In generating the draft sequence, the order of bases in each chromosome was determined at least four or five times to ensure data accuracy and to help with contig assembly. This repeated sequencing is known as genome ‘depth of coverage’. Draft sequence data is mostly in the form of10 000 bp contigs whose approximate chromosomal locations are known. The human genome sequence does not represent an exact match for any single person. DNA samples, from both male sperm and female blood, from several different anonymous individuals comprise the genomic libraries that were used for sequencing. A large number of people have donated samples for the construction of these libraries, but only a few samples have been used and neither the donors nor those responsible for the sequencing know whose DNA is actually in them. It is likely that DNA from 10 –20 individuals has been used to produce the draft genome sequence. The initial analysis of the draft sequence data was published in February 2001 by two competing groups – the HGP (Lander et al., 2001) and the biotechnology company Celera Genomics (Venter et al., 2001). HGP is a consortium of 16 academic research laboratories and institutes that used a hierarchical shotgun approach to sequencing the genome. As a publicly funded group, the data obtained from their sequencing efforts is made freely available. The Bermuda Principle (determined during a 1997 conference in Bermuda) states that sequence assemblies of 2 kbp or larger should be automatically released to public databases within 24 hours of their generation. Celera Genomics undertook a whole genome shotgun approach to sequencing the human genome. Using large numbers of DNA sequencing machines, Celera completed the sequencing phase of their project within 9 months and then employed massive computing power to reconstruct the genome based on their data.
To generate a high-quality final sequence of the human genome, additional sequencing is needed to close gaps, reduce ambiguities and allow for only a single error every 10 000 bases, giving an accuracy of 99.9999 per cent – the agreed standard for the finished sequence. The finished version will provide
9.7 FINDING GENES |
307 |
|
|
an estimated eightto nine-fold depth of coverage for each chromosome. To date, finished sequences have been generated for the three smallest human chromosomes −20, 21 and 22. Fully sequenced genomes are, in fact, not usually complete. For example, the 56 Mbp sequence of human chromosome 22 was declared essentially complete in 1999, yet only 33.5 Mbp were sequenced (Dunham et al., 1999). Also, the 180 Mbp genome of the fruit fly Drosophila was announced as completed, although just 120 Mbp were sequenced (Adams et al., 2000). Higher-eukaryotic genomes have large regions of DNA that currently cannot be cloned or assembled. These regions include telomeres and centromeres (the ends and middle of chromosomes), as well as many chromosomal areas rich in other sequence repeats. Most of the regions that cannot be sequenced contain heterochromatic DNA, which has few genes and many repeated regions. Current efforts are aimed at sequencing euchromatic DNA – defined as gene-rich areas including both exons and introns. In the case of human chromosome 22, the sequenced 60 per cent represents 97 per cent of euchromatic DNA. Similarly, nearly all the euchromatic regions were sequenced in Drosophila. Although the goal of the human genome project is to have a complete sequence for each chromosome, obtaining a full sequence still presents a great challenge.
9.7Finding Genes
Once the DNA sequence of the genome has been obtained, various methods can be employed to identify and locate the genes that reside within it.
•Sequence inspection. Genes that code for proteins comprise an ORF consisting of a series of triplet codons that specify the amino acid sequence of the protein. The ORF begins with an initiation codon (usually ATG) and ends with a termination codon (either TAA, TAG or TGA). The average length of an ORF varies between species. In E. coli, the average ORF length is 317 codons, while in yeast the average length is 483 codons. Most ORFs are, however, more than 50 codons in length. The search for a gene can therefore be thought of as a scan for an initiation and termination codon that are separated by, say, at least 100 codons. The approach works well for compact prokaryotic genomes, where genes are separated by only small regions of non-coding DNA, but is difficult to apply to the genomes of higher eukaryotes, where ORFs are split into introns and exons. Several computer programmes are available for the identification of ORFs (Fickett, 1996). In general these programmes are species specific; e.g. GRAIL (Roberts, 1991) and GeneMark (Isono, McIninch and Borodovsky, 1994)