

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf5.1 GENOMIC LIBRARIES |
189 |
|
|
cleavage only within the linker molecules which are the only ones that contain non-methylated EcoRI restriction enzyme recognition sequences. The resulting DNA fragments can then be cloned into the EcoRI restriction site of a suitable vector.
•Restriction enzymes that generate sticky ends. The genomic DNA may be initially digested with a commonly occurring restriction enzyme that
generates sticky ends. For example, digestion on genomic DNA with the restriction enzyme Sau3AI (recognition sequence 5 -GATC-3 ) generates
DNA fragments that are compatible with the sticky end produced by BamHI (recognition sequence 5 -GGATCC-3 ) cleavage of a vector. The ease of this second approach makes its use far more prevalent.
Once the DNA fragments are produced, there are cloned into a suitable vector. Often this will be a λ based vector but, as we have seen in Chapter 3, a variety of vectors are available for cloning large DNA fragments. The recombinant vector and insert combinations are then grown in E. coli such that a single bacterial colony or viral plaque arises from the ligation of a single genomic DNA fragment into the vector. E. coli cells infected with either a λ phage or transformed with a plasmid DNA are unable to support the replication of additional DNA molecules of the same type. Consequently, each λ plaque or bacterial colony contains multiple copies of the same recombinant DNA molecule. A library of these molecules is produced by pooling colonies or plaques such that sufficient are present to ensure that each genomic DNA fragment is represented at least once within the library. The main advantage of cloning large DNA fragments is that fewer individual clones must be pooled together to form a representative library. A pertinent question to ask here is how many individual colonies or plaques must be pooled to ensure that a library is truly representative of the genomic DNA from which it was made. The answer to this depends upon both the size of the genome from which the library is made and upon the average size of the cloned DNA fragments within the library. For example, if a library of the E. coli genome (4.6 Mbp) were constructed containing 5 kbp fragments, then the fraction of the genome size compared to the average individual cloned fragment size (f ) would give the lowest possible number of clones that the library must contain:
f = |
genome size |
= |
4600 000 bp |
= 920 |
fragment size |
5000 bp |
Therefore, an E. coli genomic library of this size would require at least 920 independent clones. Using the same calculation, a human genomic library containing similar sized inserts would require at least 580 000 independent

5.2 cDNA LIBRARIES |
191 |
|
|
plated out once more, and the resulting progeny collected to form an amplified library. The amplified library usually has a much larger volume than the primary library, and consequently may be screened many, or even hundreds, of times. Pooled collections of λ phages can be stored almost indefinitely. Bacterial cells harbouring plasmids are more difficult to store and there is often a high degree of recombinant clone loss upon resurrection of frozen bacterial cells. Amplification of the library is essential if the library is to be screened multiple times. However, it is possible that the amplification process will result in the composition of the amplified library not truly reflecting the primary one. As we have already discussed, certain DNA sequences may be relatively toxic to E. coli cells; as a consequence bacteria harbouring such clones will grow more slowly than other bacteria harbouring DNA sequences that do not affect bacterial growth. Such problematic DNA sequences may be present in the primary library, but will be lost, or under-represented, after the growth phase required to produce the amplified library.
5.2cDNA Libraries
Not only are the genomes of higher-eukaryotic organisms big, but also only a small fraction of the DNA contained within them codes for genes. The Human Genome Sequencing Project (Chapter 9) has estimated that genes constitute only about 1.5 per cent of the DNA contained within the genome. The knowledge of the entire genome sequence is important to understand the potential of a cell, i.e. the proteins that it could potentially produce, but perhaps more important is knowledge of the protein content that individual cells actually produce. All cells within an individual organism are derived from the same genome sequence, but the way in which the genome is transcribed and translated is unique to individual cell types, and to the individual developmental stages of each cell. Although many of the genes expressed by different cell types will be the same, e.g. the genes encoding the enzymes of the TCA cycle, some will also be different, e.g. some of the genes expressed within a skin cell will be different to those of a muscle cell. These differentially expressed genes, and the proteins that they produce, define each individual cell type. Thus, the mRNA that is contained within a cell gives us a snapshot of the genes being expressed within that cell at any particular time. mRNA actually represents only a small fraction of the total RNA contained within a cell (Table 5.2).
Most eukaryotic protein coding genes are transcribed by RNA polymerase II and the resulting mRNA is usually subjected to a number of post-transcriptional modifications, including the additions of a 7-methylguanosine cap at the 5 -end, and the addition of 100 –200 adenine residues (a poly(A) tail) at the 3 -end

5.2 cDNA LIBRARIES |
193 |
|
|
all other DNA polymerases, catalyses the addition of new nucleotides to a growing chain in a 5 to 3 direction. Reverse transcriptases generally have two types of enzymatic activity.
•DNA polymerase activity. In the retroviral life cycle, reverse transcriptase produces a DNA copy from RNA only but, as used in the laboratory, it will transcribe both single-stranded RNA and single-stranded DNA templates with essentially the same efficiency. In both cases, an RNA or DNA primer is required to initiate synthesis.
•RNaseH activity. RNaseH is a ribonuclease that degrades the RNA from RNA –DNA hybrids, such as those formed during reverse transcription of an RNA template. RNaseH functions as both an endonuclease and exonuclease to hydrolyse its target molecules.
All retroviruses encode their own reverse transcriptase (RT), but the commercially available enzymes used in cDNA library construction are derived either from Moloney murine leukemia virus (MMLV-RT) or from Avian myeloblastosis virus (AMV-RT), after purification of the enzyme from virally infected cells or following expression in and purification from E. coli. Both enzymes have the same fundamental activities, but differ in a number of characteristics, including temperature and pH optima. MMLV-RT is a single polypeptide of 71 kDa in size, while AMV-RT is composed of two polypeptide chains 64 kDa and 96 kDa in size. Most importantly, MMLV-RT has a very weak RNaseH activity compared to AMV-RT, which gives it an obvious advantage when being used to synthesize DNA from long RNA molecules.
The process of producing a double-stranded cDNA copy of an mRNA molecule is shown in Figure 5.4. The presence of a polyA tail is unique to mRNA, and provides a mechanism of distinguishing and isolating mRNA from the more abundant rRNA and tRNA molecules. mRNA can be physically isolated from its more abundant relatives by passing total RNA over a column to which polymers of deoxythymidine (oligo-dT) are bound. RNA molecules that do not contain multiple adenine residues will be unable to adhere to such a column and will flow straight through the column. mRNA molecules, on the other hand, will bind through complementary base pairing to the column and will be eluted only when the concentration of salt flowing through the column is lowered.
The cloning of cDNA is initiated by mixing short (12 –18 base) oligonucleotides of dT with purified mRNA such that the oligonucleotide will anneal to the polyA tail of the RNA molecule. Reverse transcriptase is then added and uses the oligo-dT as a primer to synthesize a single strand of cDNA in the presence of
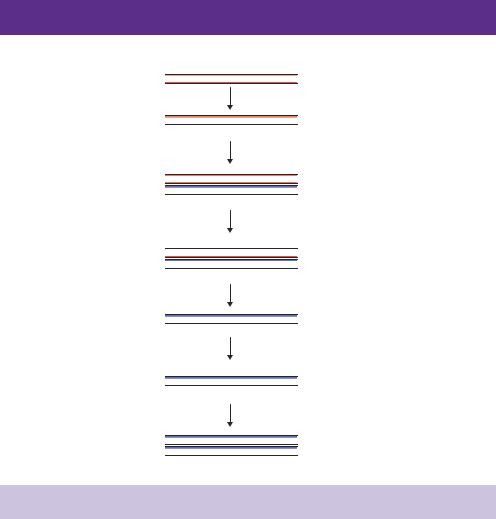
194 CLONING A GENE 5
mRNA
5'– 
 AAAAA–3'
AAAAA–3'
Oligo-dT
5'– 
 AAAAA–3' 3'–TTTTT–5'
AAAAA–3' 3'–TTTTT–5'
Reverse transcriptase + dNTPs
mRNA
5'– 
 AAAAA–3'
AAAAA–3'
3'– 
 TTTTT–5' cDNA
TTTTT–5' cDNA
Terminal transferase + dCTP
mRNA
5'– 
 AAAAACCC–3' 3'–CCC
AAAAACCC–3' 3'–CCC 
 TTTTT–5'
TTTTT–5'
cDNA
Alkaline sucrose gradient
3'–CCC 
 TTTTT–5' cDNA
TTTTT–5' cDNA
Oligo-dG
5'–GGG–3'
3'–CCC 
 TTTTT–5' cDNA
TTTTT–5' cDNA
Reverse transcriptase + dNTPs
5'–GGG 
 AAAAA–3' 3'–CCC
AAAAA–3' 3'–CCC 
 TTTTT–5'
TTTTT–5'
Double-stranded cDNA
Figure 5.4. The construction of a cDNA library. See the text for details
the four deoxynucleotide triphosphates (dNTPs). The resulting molecules will be double-stranded hybrids of one cDNA and one mRNA molecule. An oligodT primer used to make a cDNA strand will have heterologous ends. The primer can pair at numerous positions throughout the polyA tail and consequently will yield cDNA fragments of different lengths which may have been derived from the same mRNA molecule. To overcome this problem, anchored oligodT primers are often employed. In addition to the 12 –18 base dT sequence, anchored primers are constructed such that the extreme 3 -end contains either
aG, A, or C residue (Liang and Pardee, 1992). Such primers (5 -T12 – 18V-3 , where V = G, A, or C) will only efficiently initiate DNA replication if they are paired at the extreme 5 -end of the polyA tail, when the G, A, or C residue can base pair with the nucleotide immediately preceding the polyA sequence.
The production of the second DNA strand, like all DNA replication, requires
aprimer to initiate DNA synthesis. However, beyond the polyA tail, mRNA
5.2 cDNA LIBRARIES |
195 |
|
|
molecules produced from different genes will be different. Therefore, a mechanism is required to initiate DNA synthesis at sequences corresponding to the 5 -end of the mRNA. Early cDNA cloning strategies involved the formation of a hair-pin in the newly synthesized cDNA strand, which would serve as a selfpriming structure for the formation of the second strand. The hair-pin would be subsequently removed from the double-stranded cDNA by treatment with S1 nuclease (Efstratiadis et al., 1976). However, such methods invariably resulted in the loss of sequences at the 5 -end of genes, and so the second DNA strand is usually synthesized following either nick translation or homopolymer tailing.
•Nick translation. RNAse H is used to partially digest the RNA component of the RNA –DNA hybrids (Gubler and Hoffman, 1983). The remaining RNA is then used as a primer for fresh DNA synthesis using DNA polymerase I in the presence of the four dNTPs and finally DNA ligase is used to seal any remaining nicks in the DNA backbone. The resulting double-stranded cDNA molecule can subsequently be cloned into a suitable vector.
•Homopolymer tailing. The RNA –DNA hybrids formed after the first cDNA strand synthesis are treated with the enzyme terminal transferase in the presence of a single deoxynucleotide triphosphate. Terminal
deoxynucleotidal transferase (TdT) is a template independent polymerase that catalyses the addition of deoxynucleotides to the 3 -ends of DNA molecules (Chang and Bollum, 1986). TdT activity was initially identified by the analysis of immunoglobin (VDJ) recombination in which extra nucleotides were found to be inserted into the joined segments that were not present in either segment before joining (Alt and Baltimore, 1982). TdT is found at high concentration in the thymus and bone marrow where such recombination events occur, but is commercially available as a recombinant protein over-produced in and purified from E. coli. DNA (and RNA)
molecules incubated with TdT in the presence of dCTP will have multiple C residues added to their 3 -ends (Figure 5.4). Prior to the synthesis of the second DNA strand, the RNA of the RNA –DNA hybrids must be removed to provide a single-stranded template for new DNA synthesis. This can be achieved easily by treating the hybrids with alkali. RNA is hydrolysed into ribonucleotides around pH 11, while DNA is resistant to hydrolysis up to about pH 13 (Watson and Yamazaki, 1973). Increasing the pH to about 12 therefore results in the hydrolysis of the RNA, but not the DNA. Full-length cDNA strands are separated from the ribonucleotides on the basis of their
size using sucrose gradient centrifugation. The resulting cDNA strands will have multiple C residues at their 3 -ends and multiple T residues at their 5 -ends (Figure 5.4). Second-strand cDNA synthesis is then initiated using
196 |
CLONING A GENE 5 |
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
mRNA |
|||
|
5'– |
|
|
|
|
AAAAA–3' |
|
|
|
|
|
|
|||
|
|
|
|
|
cDNA synthesis |
||
|
|
|
|
|
|||
|
5'– |
|
|
|
|
|
AAAAA–3' |
|
|
|
|
|
|
||
|
3'– |
|
|
|
|
|
TTTTT–5' |
|
|
|
|
|
|
||
|
|
|
Double-stranded cDNA |
||||
|
|
|
|
|
Cloning |
||
|
|
|
|
|
|||
Promoter |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
TTTTT |
|
|
|
|
|
|
|
AAAAA |
|
|
|
|
|
|
|
|
|
|
|
|
Promoter |
|
or |
|||||
|
|
|
|
|
|||
AAAAA
TTTTT
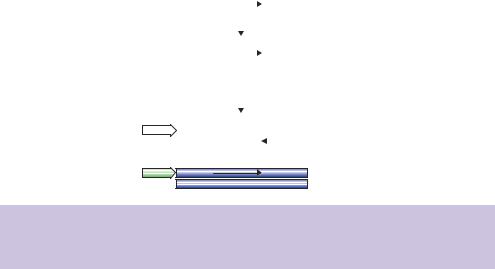
Figure 5.5. cDNA that is to be expressed must be cloned in a defined orientation so that the promoter element to which it is attached will initiate the transcription of the sense strand of the DNA, rather than the antisense strand
an oligo-dG primer that will bind, through complementary base pairing, to the newly formed polyC sequence. Reverse transcriptase, performing the role of a DNA-dependent DNA polymerase, in the presence of the four dNTPs will produce the second cDNA strand.
Homopolymer tailing has an additional advantage in that both the 5 - and 3 - ends of the original mRNA are tagged with specific and known sequences in the resulting double-stranded cDNA. This can be immensely helpful when cloning cDNA fragments in a specific orientation is required, e.g. during the expression of the cDNA. mRNA molecules are directional. The 5 -end represents the beginning of the gene sequence, and the 3 polyA tail occurs at the end of the gene sequence. Therefore, if we want to express the cDNA in, for instance, bacterial cells, it is important to ensure that only the sense strand of the cDNA is transcribed. If the antisense strand is cloned downstream of a bacterial promoter, then the resulting transcript (if produced at all) will not encode the intended protein (Figure 5.5).
5.3Directional cDNA Cloning
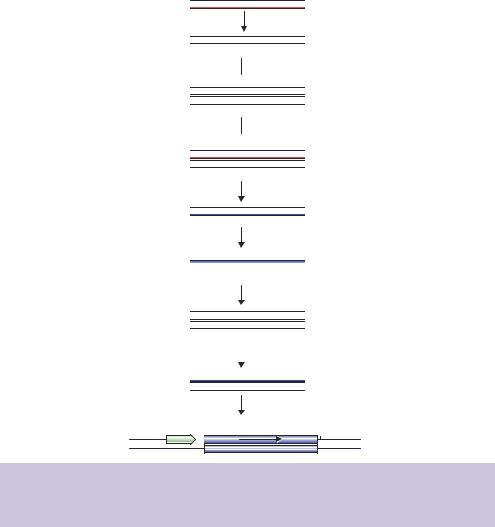
The synthesis of cDNA using modified oligonucleotides to initiate each strand of DNA synthesis allows the insertion of unique restriction enzyme recognition sites at either end of the cDNA so that cloning of the cDNA fragments can only occur in one direction (Figure 5.6). In the example shown here, the oligo-dT primer also contains additional sequences at the 5 -end that encode a XhoI
5.3 DIRECTIONAL cDNA CLONING |
197 |
|
|
mRNA
5'–

 AAAAA–3'
AAAAA–3'
Primer - 5'–GGGCTCGAGTTTTT–3'
5'–

 AAAAA-3' 3'–TTTTTGAGCTCGGG–5'
AAAAA-3' 3'–TTTTTGAGCTCGGG–5'
Reverse transcriptase
mRNA  + dNTPs
+ dNTPs
5'–

 AAAAA–3'
AAAAA–3'
3'–
 TTTTTGAGCTCGGG–5' cDNA
TTTTTGAGCTCGGG–5' cDNA
Terminal transferase
 + dCTP
+ dCTP
mRNA
5'– 

 AAAAACCC–3'
AAAAACCC–3'
3'–CCCCC
 TTTTTGAGCTCGGG–5' cDNA
TTTTTGAGCTCGGG–5' cDNA
Alkaline sucrose gradient
3'–CCCCC
 TTTTTGAGCTCGGG–5' cDNA
TTTTTGAGCTCGGG–5' cDNA
Primer - 5'–GGGGAATTCGGGGG–3'
5'–GGGGAATTCGGGGG–3'
3'–CCCCC 
 TTTTTGAGCTCGGG–5' cDNA
TTTTTGAGCTCGGG–5' cDNA
Reverse transcriptase + dNTPs
5'–GGGGAATTCGGGGG 

 AAAAACTCGAGCCC–3' 3'–CCCCTTAAGCCCCC
AAAAACTCGAGCCC–3' 3'–CCCCTTAAGCCCCC 
 TTTTTGAGCTCGGG–5'
TTTTTGAGCTCGGG–5'
EcoRI |
Double-stranded cDNA |
XhoI |
|
|
|
Cut with EcoRI and XhoI |
|
|
|
||
|
|
|
|
5'–AATTCGGGGG 

 AAAAAC–3' 3'–GCCCCC
AAAAAC–3' 3'–GCCCCC 
 TTTTTGAGCT–5'
TTTTTGAGCT–5'
Clone into EcoRI- XhoI-cut vector
EcoRI |
XhoI |
Promoter 
Figure 5.6. Directional cDNA cloning. Modified primers initiate DNA synthesis and result in the insertion of restriction enzyme recognition sequences at the 5 - and 3 -ends of the cDNA
restriction enzyme recognition site (5 -CTCGAG-3 ). As we discussed for PCR in Chapter 4, the primer initiates DNA synthesis and is itself incorporated into the extended product. Thus a XhoI restriction enzyme recognition site will be incorporated into the 3 -end of the cDNA. Similarly, the primer used to initiate the second cDNA strand contains, in addition to the oligo-dG sequence, an EcoRI restriction enzyme recognition site (5 -GAATTC-3 ) at its 5 -end. Consequently, the produced cDNA will contain an EcoRI site its 5 -end and a XhoI site at its 3 -end. The placement of these sites means that the cDNA can be cloned directionally. A plasmid bearing a suitable promoter followed by, in order, an EcoRI and a XhoI restriction enzyme recognition site will accept the