

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf78 |
BASIC TECHNIQUES IN GENE ANALYSIS |
2 |
|
|
|
(a) |
NH2 |
NH2 |
|
N |
N |
|
N |
N |
|
ATP |
PPi |
|
|
|
|
O |
|
O |
O |
|
|
N |
|
N |
|||||||||||
Lys-NH2 + −O |
|
|
|
O |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
||
|
P |
|
|
P |
|
O |
|
P |
|
|
|
|
|
O |
|||||||
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
O− |
|
O− |
O− |
|
H |
H |
|
|
|||||||||||
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|||||||||||||||
DNA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
H |
|
H |
|||||||||
ligase |
|
|
|
|
|
|
|
|
|
|
|
OH |
OH |
||||||||
(b) 5′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
5′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
C A T |
A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
G |
T A |
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
G T A T |
|
|
|
|
|
O |
|
|
|
|
C A T |
|
|
|
|
|
|
|||||||||||
3′ |
|
O |
|
|
|
|
O− |
HO |
|
|
|
5′ |
|
|
|
3′ |
|||||||||||||
|
|
|
|
P |
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
NH2 |
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
O− |
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
+ |
|
|
|
|
|
N |
|
|
|
|
|
|
N |
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
H |
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
DNA |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
ligase |
||||||||||||||
|
Lys |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
||||||||
|
|
N+ |
|
|
P |
|
|
O |
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
O− |
|
H |
H |
|
|
|
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
|
DNA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
H |
OH |
|
H |
|
|
|
|
|
|
||||||||||
|
ligase |
|
|
|
|
|
|
|
|
|
|
|
OH |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
|
|
|
H |
O |
|
|
|
|
|
|
|
N |
||||||||||||
|
|
|
Lys |
|
N+ |
|
|
|
O |
|
|
|
N |
|
|
|
|
|
|
||||||||
|
|
|
|
|
P |
|
|
|
|
|
|
O |
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
O− |
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
H |
|
|
H |
H |
|
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
DNA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
H |
OH |
|
|
H |
|||||||||||||||
|
|
ligase |
|
|
|
|
|
|
|
|
OH |
||||||||||||||||
|
|
|
|
|
Enzyme-adenylate |
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
complex |
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
C |
A |
T |
A |
|
|
|
|
|
|
|
|
|
|
|
|
G |
T |
|
A |
|||||||
|
G |
T |
A |
T |
|
|
|
O |
|
|
|
|
C |
A |
|
T |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
|
|
|
|
|
O |
|
P |
|
O− |
HO |
|
|
|
|
|
|
|
|
5′ |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
NH2
O
N
HN
Lys |
|
|
|
O |
|
P |
|
O− |
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
N |
|
N |
||||
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
O |
|
|
|
|
O |
|||||||
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
H |
H |
|
|
||
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
H |
||||
|
|
|
|
|
|
|
|
|
|
|
OH |
OH |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
A |
T |
A |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
G |
T |
A |
||||||||
|
G |
T |
A |
T |
|
|
|
|
|
|
|
|
O |
|
|
|
C |
A |
T |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
O |
|
|
|
|
P |
|
|
O |
|
|
|
|
|
|
|
5′ |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
O− |
|
|
|
NH2 |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
N |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
AMP |
|
|
|
O |
|
|
|
|
|
|
N |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
||||||||||||
|
|
|
|
−O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
P |
|
O |
|
|
|
|
|
O |
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
O− |
|
|
H |
H |
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
H |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
OH |
OH |
||||||
Figure 2.6. The mechanism of DNA joining by DNA ligase. See the text for details. This figure is adapted from Doherty et al. (1996)
2.3The Basics of Cloning
The ability to break and rejoin DNA molecules almost at will led to the first experiments in DNA cloning in 1972 (Jackson, Symons and Berg, 1972).
2.3 THE BASICS OF CLONING |
79 |
|
|
|
|
|
CC---GA |
A |
T |
|
|||
|
T |
|
|
T |
|||||
|
A |
|
|
|
|
|
|
||
G |
|
|
--- |
|
|
|
|
||
G |
|
|
|
CT |
|
|
|
|
|
|
|
|
GG |
|
|
|
|
||
|
|
A |
|
T |
A |
A |
|||
|
T |
|
|
|
|
||||
|
C |
|
|
|
|
|
|
||
C |
|
|
|
|
|
|
|
G |
|
C
|
|
Insert |
|
Vector |
|
|
|
|
|
||
5′ |
BamHI |
|
EcoRI |
3′ |
|
G G A T C C |
G A A T T C |
||||
|
|
||||
3′ |
C C T A |
G G |
C T T A A G |
5′ |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
Cut with BamHI and EcoRI |
|
|
|
|
5′ |
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
|
|
|
G A T C C |
|
|
|
|
|
G |
|
|
′ |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
- |
|
′ |
|
|
|
|
|
|
|
|
|
|
|
|
|
G |
|
|
|
|
3′ |
G |
|
|
|
|
|
C T T A A |
5′ |
|
G-5 |
|||
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
C |
A |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
T |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
5 |
′- |
A |
|
|
|
|
|
||
|
|
A |
|
|
|
|
|
T |
|
|
|
|
T |
|
|
|
3′ |
C |
|
|
|
|
||
|
|
|
- |
|
|
|
|
G |
|
DNA ligase
|
|
C |
|
|
C |
|
|
|
T |
|
|
|
A |
|
|
G |
|
G |
|
G |
|
|
|
|
|
G |
|
|
A |
|
|
|
T |
|
|
|
C |
|
|
|
C |
|
|
G |
|
|
A |
|
|
A |
|
|
T |
|
|
|
T |
|
C |
|
C |
T |
|
|
T |
|
|
A |
|
|
A |
|
|
G |
|
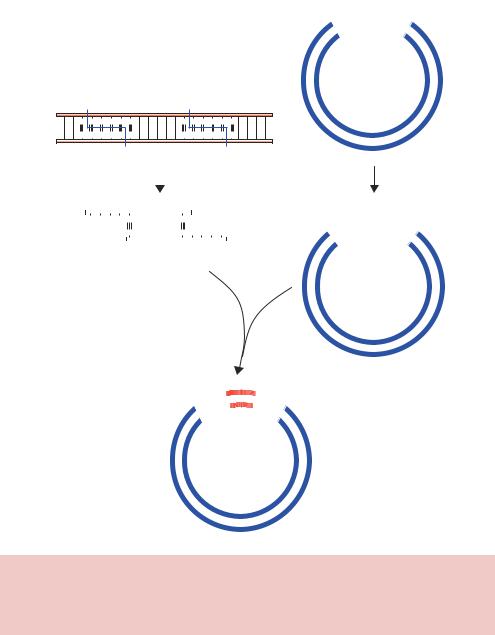
|
Figure 2.7. Breaking and joining DNA using restriction enzymes and DNA ligase. Linear DNA (insert) and a closed-circular plasmid DNA (vector) each contain the recognition site for BamHI and EcoRI. Mixing the DNA fragments with compatible ends together in the presence of DNA ligase can result in the formation of vector–insert hybrid DNA molecules
For the first time it was possible to extract a fragment of DNA from one source and insert, or clone, it into the DNA from another source. Perhaps the most common type of cloning experiment involves the insertion of a foreign piece of DNA into a suitable vector so that the foreign DNA may be propagated in E. coli. In Chapter 3 we will discuss the various different
80 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
types of vector that are available, but at this stage we could consider the vector as a closed-circular double-stranded plasmid DNA molecule. If we wish to insert foreign DNA sequences into this vector, we need to cut it to produce a linear DNA onto which we can attach other DNA sequences using DNA ligase.
Let us first consider the insertion of DNA into the vector using two different restriction enzymes (Figure 2.7). Treatment of both a vector and insert DNA sequences with the restriction enzymes will generate a number of DNA fragments. In the vector the recognition sites for the restriction enzymes are located close to each other. As we will see in Chapter 3, this is very common in engineered plasmids. Cutting such a vector with BamHI and EcoRI will yield two fragments – a large one, comprising the majority of the vector, and a small one, representing the DNA between the restriction enzyme recognition sites. In the presence of DNA ligase, neither of these fragments is able to ligate to itself because the DNA ends are not compatible with each other. Digestion of the linear insert DNA sequence with BamHI and EcoRI results in the generation of three DNA fragments. Only one of the fragments contains a BamHIand EcoRI-compatible end; the others represent the DNA at either end of the fragment. Mixing the vector DNA and insert DNA that are compatible with each other will result in the formation of hydrogen bonds between the two DNA molecules. If the ends were not compatible, this hydrogen bonding would not occur. The addition of DNA ligase to the hydrogen bonded intermediate will result in the sealing of the DNA backbone and the formation of a vector –insert hybrid DNA molecule. If one of the other insert DNA fragments becomes hydrogen bonded to the vector, say via its BamHI-compatible end, then ligation will not result in the formation of a closed-circular vector. As we will see in the next section, such DNA molecules are not replicated when they are transformed into bacteria.
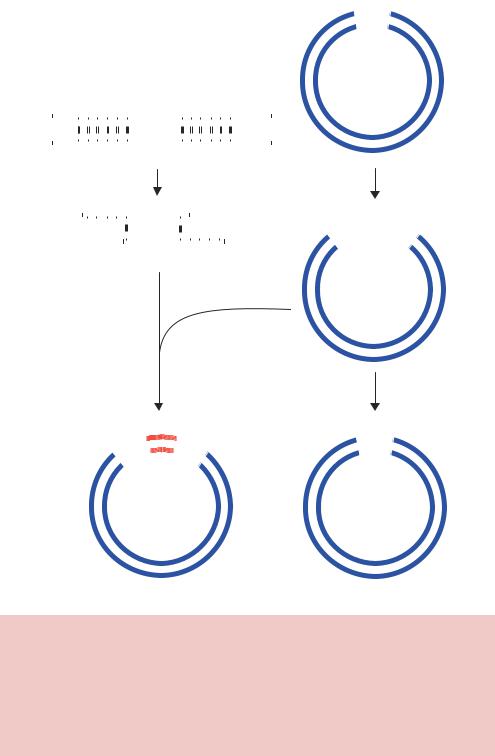
This type of cloning scheme works because the vector DNA that has been cut with the two restriction enzymes contains non-complementary DNA ends. If the vector had been cut using a single restriction enzyme, or with two restriction enzymes that left the same sticky ends, then the vector could easily recircularize in the presence of DNA ligase. Treating the vector with a phosphatase enzyme after it has been cut with the restriction enzymes, however, can prevent this. Phosphatases catalyse the removal of 5 phosphate groups from nucleic acids and nucleotide triphosphates. Since phosphatase treated DNA fragments lack the 5 phosphate required by DNA ligase, such treatment will inhibit vector self-ligation and will promote the formation of vector –insert DNA hybrids. Such a cloning scheme is shown diagrammatically in Figure 2.8. When the vector has been treated with phosphatase, DNA ligase
2.3 THE BASICS OF CLONING |
81 |
|
|
T |
|
GAA TC |
|
T |
|
CT AAG |
|
Vector
|
|
|
|
|
|
|
Insert |
||||||||||||
5′ |
|
|
|
EcoRI |
|
|
|
|
EcoRI |
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
|
G |
A A T T C |
|
|
|
|
G |
A A T T C |
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3′ |
|
|
|
C T T A A |
G |
|
|
|
|
C T T A A |
G |
|
|
|
|
|
5′ |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cut with EcoRI
5′ |
|
|
|
|
|
|
|
|
3′ |
||||
|
A A T T C |
|
|
|
|
|
|
||||||
|
|
|
|
|
|
G |
|||||||
|
|
3′ |
|
G |
|
|
|
|
C T T A A |
|
5′ |
||
|
|
|
|
|
|
|
|
|
|
||||
|
' |
|
|
|
|
3 |
|
|
5' |
- |
|
|
||
G |
|
|
A- |
|
|
|
A |
|
|
|
|
T |
|
|
|
T |
|
|
|
|
C |
|
|
|
5 |
'- |
|
|
|
A |
A |
|
|
|
T |
|
|
|
T |
|
|
3' |
C |
|
|
|
- |
|
|
|
G |
|
Treat vector with phosphatase
DNA ligase
|
|
C |
|
|
G |
A |
A |
|
|
|
T |
|
|
|
T |
|
|||
|
T |
|
|
|
|
|
T |
||
|
A |
|
|
|
|
|
|
||
A |
|
|
|
|
|
|
|
||
G |
|
|
G |
C |
|
|
|
|
C |
|
|
A |
T |
|
|
|
|
||
|
A |
|
|
T |
|
|
|
||
|
T |
|
|
|
|
A |
|
|
|
|
T |
|
|
|
|
|
A |
|
|
C |
|
|
|
|
|
|
G |
||
DNA ligase
GA |
AT |
TC |
|
CT |
T A |
AG |
Vector with insert |
Reformed vector |
Figure 2.8. The basics of cloning into a plasmid vector containing a single unique restriction enzyme recognition site. The vector contains a single EcoRI recognition site, while the insert has two. Cutting both with the enzyme generates the fragments shown. Adding ligase to the cut vector will probably result in its reformation. This can be prevented by treating the cut vector with phosphatase to remove the 5 phosphate residues from the ends of the DNA. The cut insert can provide the missing phosphate that DNA ligase requires, so mixing the vector and insert will result in the formation of hybrid DNA molecules
82 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
5′-GAATTC-3′
3′-CTTAAG-5′
Vector
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
EcoRI |
|
|
|
|
|
|
|
|
|
|
|
5′-G-OH |
PO4-AATTC-3′ |
||||
|
|
|
|
|
|
|
|
|
|
|
3′-CTTAA-PO4 |
HO-G-5′ |
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Phosphatase |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5′-G-OH |
OH-AATTC-3′ |
||||
|
|
|
|
|
|
|
|
|
|
|
3′-CTTAA-OH |
HO-G-5′ |
||||
5′PO4- |
|
|
|
|
|
|
|
|
|
|
-OH3′ |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
||||||
|
A A T T C |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
G |
|
|
Insert + DNA ligase |
|||||||
|
|
3′HO- |
|
G |
|
|
|
|
C T T A A |
|
-PO45′ |
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Breaks in DNA backbone
5′-GAATTC |
G AATTC-3′ |
3′-CTTAA G |
CTTAAG-5′ |
Transform into bacteria
5′-GAATTC GAATTC-3′
3′-CTTAAG CTTAAG-5′
Vector plus insert
Breaks repaired by bacteria
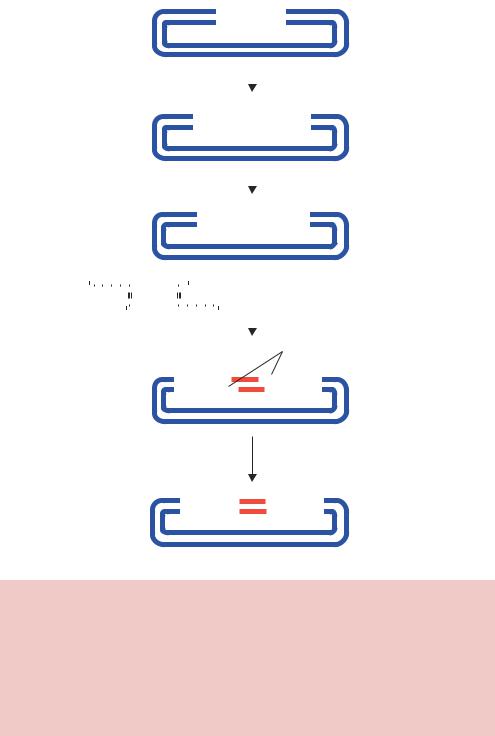
Figure 2.9. The ligation of vector DNA that has been treated with phosphatase to a compatible insert. The vector is cut with the restriction enzyme EcoRI and then treated with phosphatase to remove the phosphates from the free 5 -ends of the cut DNA. The ligation of a compatible insert into this vector will result in the ligation of only the 5 -ends of the insert with the vector. The 3 -end of the insert (a hydroxyl group) and the 5 -end of the vector (also a hydroxyl group) will be unable to ligate. Transformation of the vector–insert hybrid into bacteria, however, will result in the repair of the broken DNA strands to form the complete vector plus insert plasmid

2.3 THE BASICS OF CLONING |
83 |
|
|
will be able to seal the nicks in the DNA phosphodiester backbone on one strand only (Figure 2.9). However, once these molecules are transformed into bacteria, the break on the other strand is repaired using the bacterial DNA repair systems.
In cloning DNA fragments, there are of course many occasions when restriction enzyme recognition sites either do not occur or do not occur in the correct place within a fragment you are try to clone. This problem can be overcome in a number of ways.
•Clone into a blunt-ended restriction site. If the restriction enzyme in the vector leaves blunt ends (like EcoRV shown in Figure 2.5) then any other blunt-ended DNA fragment can be ligated into the cut vector. A number of restriction enzymes give rise to blunt ends after cutting DNA. Other sites can be made blunt by either cleaving off the overhanging ends with a nuclease (e.g. mung bean nuclease) or by ‘filling in’ the overhanging ends using a DNA polymerase. Such fill-in reactions are often performed with the Klenow fragment of DNA polymerase I in the presence of the appropriate deoxynucleotides. For example, filling in the ends of EcoRI cut DNA (Figure 2.5) would require both dATP and dTTP, and filling the ends of BamHI cut DNA would require all four deoxynucleotide triphosphates. The major drawback to blunt-end cloning is the inefficiency of DNA ligase at carrying out these reactions.
•Using oligonucleotide linkers. If you want to join DNA fragments together that have, say, EcoRI and PstI ends respectively, you can synthesize a small synthetic DNA molecule to link the two ends together. DNA ligase will then be able to efficiently seal the two non-compatible sticky ends by using the linker as a bridge between the two.
Single-stranded linker:
EcoRI |
Linker |
PstI |
|
5′-G |
5′-AATTTGCA-3′ |
G-3′ |
|
3′-CTTAA |
|
|
ACGTC -5′ |
|
DNA ligase |
|
|
|
|
|
|
|
5′-GAATTTGCAG-3′ |
|
|
|
3′-CTTAAACGTC- 5′ |
|
|
84 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
|
|
|
|
Double-stranded linker: |
|
|
|
|
EcoRI |
Linker |
BamHI |
|
5′-G |
5′-AATTAGATCA -3′ |
GATCC-3′ |
|
3′-CTTAA |
3′-TCTAGTCTAG -5′ |
G-5′ |
DNA ligase
5′-GAATTAGATCAGATCC-3′
3′-CTTAATCTAGTCATGG- 5′
•Mutagenesis to create new restriction sites. The sequence of the DNA may be altered to create or destroy restriction sites. We will discuss this further in Chapter 7.
Now that it was possible to construct hybrid DNA molecules, the next problem was to try to get these hybrid DNA molecules into living cells so that the DNA could be replicated and the genes for which they code could be expressed.
2.4Bacterial Transformation
Before 1970, there had been many attempts to transform E. coli cells with foreign DNA. In general, however, little progress could be made. Going back to the experiments of Griffith and Avery, MacLeod and McCarty (Chapter 1), we know that transformation of some bacteria will occur with naked DNA, but it is a rare event that occurs at low frequency. Additionally, as we have already seen, bacteriophages can efficiently infect various strains of E. coli, but if the same experiment is performed with naked DNA, the efficiency of transformation is very low. There are several reasons for this.
(a)Getting naked DNA into cells is not a trivial problem. DNA is highly charged and will not easily pass through the membranes that surround the bacterium. In the early 1970s, however, methods were devised to make E. coli cells competent for the uptake of naked DNA – such methods are discussed below.
(b)What is the fate of the foreign DNA once it enters the cell? For the foreign DNA to be maintained and replicated with the bacterium, it must either be integrated into the bacterial chromosome, so that it will be subsequently propagated as part of the bacterial genome, or be independently replicated. The exact mechanism whereby integration occurs is not clear and it is
2.4 BACTERIAL TRANSFORMATION |
85 |
|
|
usually a rare event. If the foreign DNA fails to be integrated, it will probably be lost during growth of the bacterial cells. The reason for this is straightforward; in order to be replicated DNA molecules must contain an origin of replication. Fragments of DNA lacking an origin of replication – even if they survive the bacterial restriction systems – will be diluted out of the host cells after cell division and will eventually be lost. Even if a foreign DNA molecule contains an origin of replication, this may not function in the bacterial cells into which the DNA has been transformed. If fragments of DNA are not able to independently replicate, the obvious solution is to attach them to a suitable replicon. Such replicons are known as vectors or cloning vehicles. Small plasmids and bacteriophages are the most convenient vectors since they are replicons in their own right, maintenance does not necessarily require integration into the host genome and their DNA can be readily isolated in an intact form. The different plasmids and bacteriophages that are used as vectors are described detail in Chapter 3.
(c)Monitoring the transformation process. Assuming that you are able to get foreign DNA into a bacterial cell and have it stably maintained, how can you distinguish the transformed cells from those that have not been transformed? Even if the foreign DNA encodes a gene product, the differences between prokaryotic and eukaryotic gene expression (Chapter 1) mean that it would be unlikely that foreign genes would be efficiently transcribed and translated in the transformed cells. The solution to this problem is to insert the foreign DNA into a cloning vector that also contains a selectable marker that will be expressed in the transformed cells. These markers, usually antibiotic resistance genes, will be discussed in more detail in Chapter 3.
In 1970, it was found that treating E. coli cells with calcium chloride (CaCl2) allowed them to take up naked bacteriophage DNA (Mandel and Higa, 1970). This chemical transformation treatment was also subsequently shown to allow plasmids to enter bacterial cells, at varying levels of efficiency. Increased transformation efficiencies have been observed using high voltage electric pulses in a process called electroporation, and using a gene gun. The process of transformation results in the insertion of a DNA molecule into the host cell. All commonly used plasmid and bacteriophage vectors used to clone foreign DNA fragments allow for the insertion of a single vector molecule into the host cell. This single molecule may be amplified many times within the host, but all of the resulting molecules are identical. A consequence of this is that if a mixed population of DNA fragments is ligated into a common vector and transformed

86 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
into, say, E. coli, then the resulting bacterial colonies will each contain one, and only one, type of recombinant DNA molecule. The mixed population of DNA fragments is segregated into its individual components during the transformation and cell growth processes. This is particularly important in the isolation of single recombinant DNA species from complex DNA libraries (Chapter 5).
2.4.1 Chemical Transformation
Chemical transformation of E. coli cells is a simple process. Essentially, the cells are grown to mid-log phase, harvested by centrifugation and resuspended in a solution of calcium chloride. The foreign DNA – often contained within a plasmid – and the now competent cells are then incubated on ice and subsequently subjected to a brief (30 s) heat shock at 37 –45 ◦C. Nutrient medium is then added to the cells and they are allowed to grow for a single generation to allow the phenotypic properties conferred by the plasmid (e.g. antibiotic resistance) to be expressed. Finally, the cells are plated out onto a selective medium such that only cells that have taken up the foreign DNA will grow. The role of calcium chloride in this process is not clear. It is thought to affect the bacterial cell wall, and may also be responsible for binding DNA to the cell surface. The actual uptake of DNA is thought to be stimulated by the brief heat shock (Figure 2.10).
(a) |
Heat |
CaCl2 |
shock |
(b) |
Pores |
|
reseal |
(c)
DNA-coated |
DNA and bead |
bead |
dissociate |
Figure 2.10. Three methods for the transformation of cells. a) Chemical transformation. Treatment of cells with calcium ions can make cells competent for the uptake of DNA. The DNA may adhere to the surface of the cell and uptake is mediated by a pulsed heat-shock. b) Electroporation. Cells are treated with an electrical pulse, which mediates the formation of pores. DNA can enter the cell before the pores spontaneously reseal. c) The gene gun. DNA molecules bound to a bead are fired at cells and are able to enter the cytoplasm. Here, the bead and the DNA dissociate
2.4 BACTERIAL TRANSFORMATION |
87 |
|
|
Since the transformation of E. coli is an essential step in many cloning experiments, the process should be as efficient as possible. The efficiency of transformation is governed by a number of host-specific and other factors, but the molecular processes by which transformation occurs are not well understood, and conditions by which efficient transformation can take place are determined empirically. Transformation efficiencies are usually increased if
•the bacterial cells to be transformed are derived from strains that are deficient in restriction systems to reduce the likelihood of degrading the foreign DNA,
•certain exonucleases (e.g. recBC) are mutated in the E. coli host cell and
• the competent E. coli cells are treated not just with calcium ions, but also with a variety of other divalent cations (e.g. rubidium and manganese) (Hanahan, 1983).
This chemical transformation procedure is applicable to most E. coli K12 strains, with typical transformation efficiencies of 107 –109 transformants per microgram of DNA added being achieved, depending on the particular strain of E. coli being employed (Liu and Rashidbaigi, 1990).
2.4.2Electroporation
Electroporation is the use of an electric field pulse to induce microscopic pores within a biological membrane. These pores, called ‘electropores’, allow molecules, ions and water to pass from one side of the membrane to the other. If a suitable electric field pulse is applied, then the electroporated cells can recover, with the electropores resealing spontaneously, and the cells can continue to grow. Pore formation is extremely rapid (approximately 1 µs), while pore resealing is much slower, and is measured in the order of minutes. The use of electroporation to transform both bacterial and higher cells became very popular throughout the 1980s. The mechanism by which electroporation occurs is not well understood and hence, like chemical transformation, the development of protocols for particular applications has usually been achieved empirically by adjusting electric pulse parameters (amplitude, duration, number and inter-pulse interval) (Ho and Mittal, 1996; Canatella et al., 2001).
Two main factors seem to influence the formation of electropores – the types of cell that are used, and the amplitude and duration of the electric pulse that is applied to them. Certain cell types respond well to this type of treatment while others are more refractory – in general, however, most cells can take