

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf88 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
up DNA when they are electroporated with varying degrees of efficiency. The pulse amplitude and duration are critical if electropores are to be induced in a particular cell. The product of the pulse amplitude and duration has to be above a lower limit threshold before pores will form, beyond which the number of pores and the pore diameter increase with the product of amplitude and duration. An upper limit threshold is eventually reached, at high amplitudes and durations, when the pore diameter and total pore area are too large for the cell to repair. The result is irreversible damage to the cell. During the electroporation pulse, the electric field causes electrical current to flow through the cells that are to be transformed. Buffers and bacterial growth media contain ionic species (e.g. Na+) at concentrations high enough to cause high electric currents to flow. These currents can lead to dramatic heating of the cells that can result in cell death. Heating effects are consequently minimized by using a relatively high-amplitude, short-duration pulse or by using two very short-duration pulses (Sukharev et al., 1992). Additionally, the cells to be electroporated are extensively washed in distilled water to remove any traces of salt that could ‘spark’ when the pulse is applied to them.
2.4.3Gene Gun
The gene gun is a device that literally fires DNA into target cells (Johnston and Tang, 1994). The DNA to be transformed into the cells is coated onto microscopic beads made of either gold or tungsten. The coated beads are then attached to the end of a plastic bullet and loaded into the firing chamber of the gene gun. An explosive force fires the bullet down the barrel of the gun towards the target cells that lie just beyond the end of the barrel. When the bullet reaches the end of the barrel it is caught and stopped, but the DNA-coated beads continue on towards the target cells. Some of the beads pass through the cell wall and into the cytoplasm of the target cells. Here, the bead and the DNA dissociate and the cells become transformed. The gene gun is particularly useful for transforming cells that are difficult to transform by other methods, e.g. plant cells. It is also gaining in use as a method for transferring DNA constructs into whole animals. For example, a vaccine has been developed against foot and mouth disease, a highly virulent viral infection of farm animals. The vaccine is composed of several viral genes that when expressed in the pig will give the animal resistance to infection by the natural virus (Benvenisti et al., 2001).
2.5Gel Electrophoresis
The progress of the first experiments on cutting and joining DNA molecules were monitored using velocity sedimentation in sucrose gradients. This type
2.5 GEL ELECTROPHORESIS |
89 |
|
|
of technique, which relies on separation based on size alone after extensive centrifugation through a tube containing high levels of sucrose, requires relatively large amounts of DNA, and is unable to distinguish small changes in the size of a DNA molecule. Separation techniques that needed less material and gave a high degree of separation were required to effectively monitor genetic engineering experiments. As we have already seen, DNA is a highly charged molecule. The phosphates that form the sugar –phosphate backbone of each DNA strand provide a high degree of negative charge. A small DNA fragment will have less negative charge than a large DNA fragment since it contains fewer phosphates. The overall charge per unit length for both a small and a large DNA molecules is, however, identical. So, if an electric current is applied to a sample of small and large DNA fragments in free solution, they will both move to the positive electrode (anode) at the same rate, assuming that friction is negligible in free solution. Therefore, a mechanism by which DNA molecules could be separated would be to increase the amount of friction so that small DNA molecules would move to the anode faster by virtue of having less friction than larger DNA molecules. Running the DNA fragments through a gel can provide the necessary friction to separate DNA fragments of different sizes.
2.5.1 Polyacrylamide Gels
Polyacrylamide gel electrophoresis (PAGE) had been introduced in 1959 as a method for separating proteins (Raymond and Weintraub, 1959). The technique is, however, equally applicable to the separation of nucleic acids. The pore size of this kind of gel may be varied, by altering the percentage polyacrylamide used to construct the gel (from 3 to 30 per cent), for separating molecules of different sizes. PAGE is a powerful technique in the analysis of DNA molecules, and is able to very effectively separate DNA molecules that differ in size by as little as a single base pair. This high level of resolution makes PAGE ideal for the analysis of DNA sequence. The technique is, however, limited to relatively small DNA molecules (less than 1000 bp in length). Large DNA molecules are unable to enter the pores of the polyacrylamide and are consequently not separated by the gel. Since most vectors that are commonly used for cloning genes are bigger than can be resolved by PAGE, an alternative technique was required.
2.5.2Agarose Gels
Agarose is a naturally occurring colloid that is extracted from seaweed. It is a linear polysaccharide made up of the basic repeat unit agarobiose, which comprises alternating units of galactose and 3,6-anhydrogalactose. Agarose
90 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
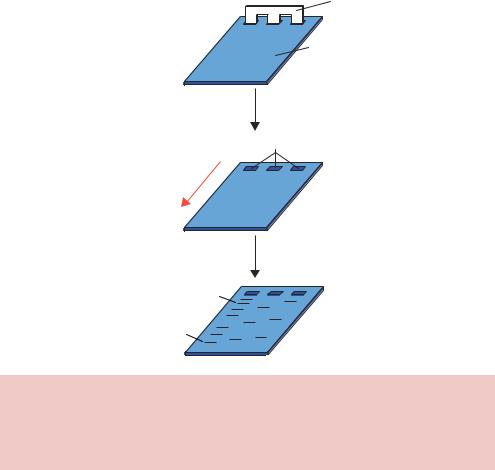
gels are formed by suspending dry agarose, at concentrations ranging between 1 and 3 per cent, in aqueous buffer, then boiling the mixture until a clear solution forms. This is poured into a suitable gel former containing a comb to form wells, and allowed to cool to room temperature to form a rigid gel (Figure 2.11). After the gel has set, the comb is removed and samples of DNA can be loaded into the resultant wells. The gel is then subjected to a constant electric field (in the range of 10 V/cm gel) and the DNA will migrate toward the positive electrode (anode).
The resolution of an agarose gel is inferior to that obtained by PAGE. The bands formed in an agarose gels are relatively fuzzy because the pore size cannot be accurately controlled. A 1 per cent agarose gel contains a wide variety of pore sizes, while a 2 per cent gel on average contains smaller pores but these are still widely variable. Once DNA fragments have been
Comb
Agarose gel
DNA in wells
−
Direction of DNA migration
+
Electrophoresis
Large DNA fragment
Small DNA fragment
Figure 2.11. Agarose gel electrophoresis. Molten agarose is poured into a former containing a comb. After the gel has set, the comb is removed to form wells into which DNA samples may be loaded. An electric current is the applied to move the negatively charged DNA through the gel towards the positive electrode. Smaller DNA molecules generally run through the gel with a higher mobility than larger DNA fragments
2.5 GEL ELECTROPHORESIS |
91 |
|
|
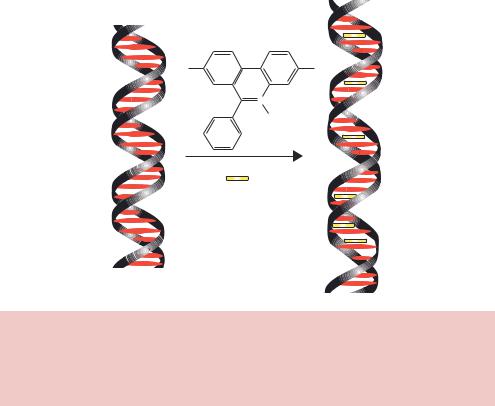
separated through an agarose gel, they must be stained so that the DNA can be visualized. The most common method of staining involves soaking the gel in a solution of ethidium bromide. Ethidium bromide is a flat planar molecule that is able to intercalate between the stacked base pairs of DNA (Figure 2.12). The binding of ethidium bromide to DNA results in distortion of the doublehelical structure and localized unwinding of the helix. Ethidium bromide will bind very efficiently to double-stranded DNA, but less so to single-stranded DNA and RNA because of the relative lack of base stacking. Soaking a DNA containing gel in ethidium bromide will result in concentration of the chemical within the DNA. Illumination of the soaked gel with light in the ultraviolet range (260 – 300 nm) results in fluorescence of ethidium bromide, and the DNA shows up on the gel as a band of fluorescence.
As we have already seen, the charge per unit length of DNA is constant, and in a gel you would expect friction to increase in direct proportion to the length of DNA – a 2000 bp fragment should experience twice as much friction as a 1000 bp fragment. Therefore there should be a direct and inverse relationship between the mass of DNA and its migration rate through a gel.
H2N |
NH |
N Br+
C2H5
Ethidium Bromide
Figure 2.12. The binding of ethidium bromide to DNA. Ethidium bromide is a flat, planar molecule that is able to intercalate in between the stacked bases of doublestranded DNA. The binding of ethidium bromide distorts the double helix and increases its overall length. DNA to which ethidium bromide is bound fluoresces when viewed under ultraviolet light
92 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
Excellent separation of DNA molecules in the range of 200 –15 000 bp is achieved using agarose gels. Two main factors govern the speed at which a DNA fragment will migrate through an agarose gel when a constant electric current is applied – its molecular mass (or length) and its shape. In general small DNA fragments will migrate faster through an agarose gel than large DNA fragments (Figure 2.13(a)). However, if we look carefully at the way in which DNA fragments of a known length run through an agarose gel, we can see that there is not a direct inverse relationship between DNA fragment size and distance migrated (Figure 2.13).
(a)
−
DNA migration
Direction of
+
(b) |
Size |
Distance |
(c) |
12500 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
(bp) |
migrated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10000 |
(mm) |
|
10000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
8000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
10000 |
12.00 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6000 |
(bp) |
7500 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
4000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
5000 |
8000 |
12.85 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3500 |
6000 |
14.55 |
size |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5000 |
16.12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
3000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
2500 |
4000 |
18.24 |
Fragment |
5000 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
2000 |
3500 |
19.74 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1500 |
3000 |
21.86 |
|
2500 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2500 |
23.97 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
1000 |
2000 |
27.32 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
750 |
1500 |
31.72 |
|
|
0 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
1000 |
38.79 |
|
|
10 |
20 |
30 |
40 |
50 |
60 |
||||||||||||||||||
|
|
|
|||||||||||||||||||||||||
500 |
750 |
43.29 |
|
|
|
|
|
Distance migrated (mm) |
|
|
|
|
|||||||||||||||
500 |
50.25 |
|
|
|
|
|
|
|
|
|
|||||||||||||||||
|
250 |
59.35 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
250 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(d) |
4.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
size |
4 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
fragment |
3.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Log |
2.5 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 |
20 |
30 |
40 |
50 |
60 |
|||||||||||||||||
Distance migrated (mm)
Figure 2.13. The migration of DNA fragments through an agarose gel. (a) An agarose gel showing the separation of DNA fragments of known size. (b) The size of a DNA fragment and the distance migrated through the gel. (c) A plot of fragment size against distance migrated taken from the data shown in (b). This indicates that the relationship between fragment size and distance migrated is not linear. (d) A plot of the log of fragment size against distance migrated. This indicates that there is a direct, inverse, relationship between distance migrated and the log of the size of a DNA fragment
2.5 GEL ELECTROPHORESIS |
93 |
|
|
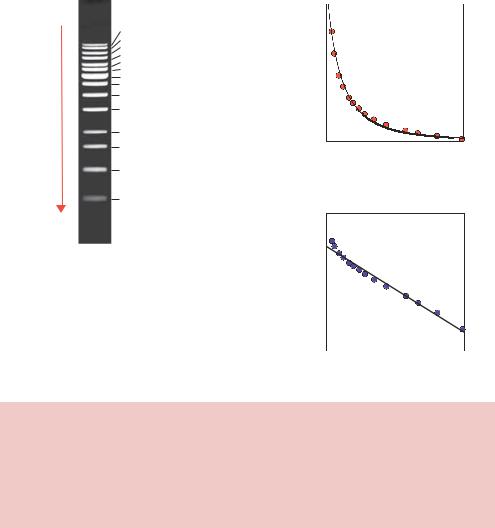
If we measure the distance that DNA molecules of a known size migrate through an agarose gel, and plot the data as size of the DNA fragment against distance migrated, we see that the migration distance is inversely proportional to the log of size (Figure 2.13(d)). This effect is most readily observed by inspection of the distances that particular DNA fragments migrate from the well after electrophoresis. DNA fragments of 500 and 1000 bp are separated widely on the gel, while DNA fragments of 8000 and 10 000 bp run close to each other on the gel (Figure 2.13(a)). A consequence of the inverse log relationship is that any DNA fragment greater than about 30 kbp will migrate in approximately the same location as much larger fragments on an agarose gel. That is to say, large DNA fragments will not be resolved from one another, and so agarose gels do not effectively separate large DNA molecules. A DNA fragment of 30 kbp runs on an agarose gel in the same place as a DNA fragment of 60 kbp. Both of these DNA fragments are able to enter the pores of the gel, and do pass through the gel driven by the electric current, but separation is not achieved. Separation of DNA fragments of this size cannot be achieved by either running the gel longer, or by lowering the concentration of agarose within the gel. To understand this phenomenon, we need to think how DNA fragments actually travel through the pores of a gel.
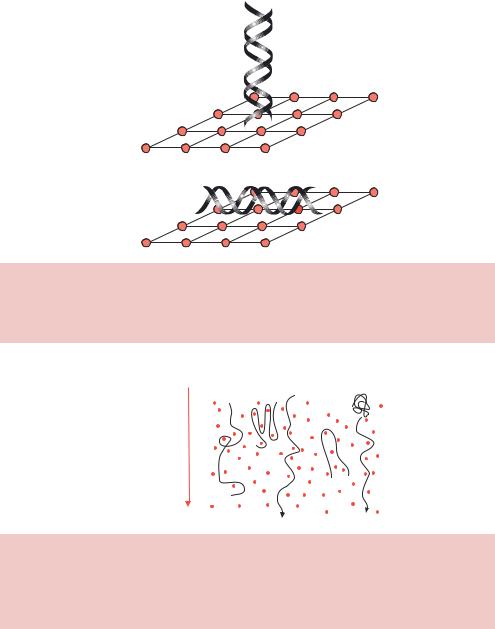
DNA is a long, thin highly charged polymer. To travel through the pores of a gel, the DNA will tend to take the path of least resistance and travel end-first through the gel pore (Figure 2.14). Several different theoretical models have been put forward to explain the movement of DNA through gels (Slater, Mayer and Drouin, 1996). Perhaps the simplest way to think about highly flexible DNA molecules travelling through a gel is to imagine them snaking (or reptating) their way through the pores of the gel matrix (Figure 2.15). Small DNA fragments will be able to pass through the gel pores more easily than longer DNA fragments due to the sieving effect of the pores. Large DNA molecules travelling in this fashion, however, may become entangled or knotted within the gel and will thus be retarded beyond the level expected based on size alone. DNA fragments above 30 000 bp in length appear to knot sufficiently to inhibit reptation through the gel pores. Larger fragments suffer the same fate, and consequently DNA fragments above 30 kbp run in the same place on an agarose gel.
The second major factor influencing migration through a gel is the topology or structure of a particular DNA fragment. For instance, plasmid DNA isolated from E. coli cells is invariably negatively supercoiled closed-circular molecules. These are relatively compact structures that run quickly through agarose gels (Figure 2.16). If one strand of the plasmid double helix becomes broken (nicked) then the supercoiling within the plasmid will be lost, and the more open structure of the relaxed plasmid will migrate more slowly through an
94 |
BASIC TECHNIQUES IN GENE ANALYSIS 2 |
|
|
DNA passes through pores
DNA trapped in pores
Figure 2.14. DNA is thought to travel through the pores of a gel in an end-on fashion. If we think of an agarose gel as a meshed network of pores, then we can imagine DNA can more readily pass through the pores if it travels end-on rather than side-on. This end-on movement is sometimes referred to as snaking or reptation
−
Direction of electrophoresis
+
Figure 2.15. DNA snaking through the pores of a gel. DNA molecules moving through the pores of a gel may become trapped in a variety of ways. Larger DNA molecules are more likely to be trapped due to their length than smaller ones. This may be the reason that DNA molecules larger than about 30 kbp all run in about the same place in a conventional agarose gel
agarose gel. If the same plasmid is treated with a restriction enzyme that cleaves it once, then this linearized DNA will run with a mobility intermediate between those of the supercoiled and the nicked molecules. Therefore, DNA molecules that all contain precisely the same number of base pairs can run in several different locations on an agarose gel depending upon the topology of the DNA.

2.5 GEL ELECTROPHORESIS |
95 |
|
|
−
Nicked
Linear
 Supercoiled
Supercoiled
+
Figure 2.16. The effect of topology on the mobility of DNA fragments. Three DNA fragments, each containing precisely the same number and sequence of base pairs, can run in different places on an agarose gel. Supercoiled DNA is highly compacted and runs rapidly through the gel. If just one of the DNA strands of supercoiled DNA becomes nicked – i.e. a single break in one strand of the sugar– phosphate backbone – then the molecule adopts an open structure with a low mobility. Linear DNA, in which the DNA backbone is broken in both strands, runs with an intermediate mobility on an agarose gel
2.5.3Pulsed-field Gel Electrophoresis
As we have seen above, all DNA fragments above about 30 kbp run with the same mobility regardless of their size. This is seen in the gel as a large diffuse band. To overcome the size limitation of resolution in an agarose gel, Schwartz and Cantor introduced pulsed-field gel electrophoresis (PFGE) as a method for resolving extremely large DNA molecules (Schwartz and Cantor, 1984). Rather than subjecting DNA fragments in a gel to a continuous static electric field, they altered the direction of the electric current to alter the path of DNA molecules as they travel through the gel (Figure 2.17). Using this technique, the upper size limit of DNA separation in agarose gels was raised from 30 –50 kbp to well over 10 Mbp (10 000 kbp).
If the DNA is forced to change direction during electrophoresis, different sized fragments within the diffuse unresolved DNA band begin to separate from each other; perhaps the changes in direction inhibits, or reduces, knot formation. With each reorientation of the electric field relative to the gel, the smaller-sized DNA fragments will begin moving in the new direction more quickly than the larger DNA fragments. Thus, the larger DNA lags behind, providing a separation from the smaller DNA. The original pulsed-field systems used the uneven electric fields generated from static electrodes. As a consequence, the DNA did not run in straight lanes, making interpretation of gels difficult. Ideally, the DNA should separate in straight lanes to simplify
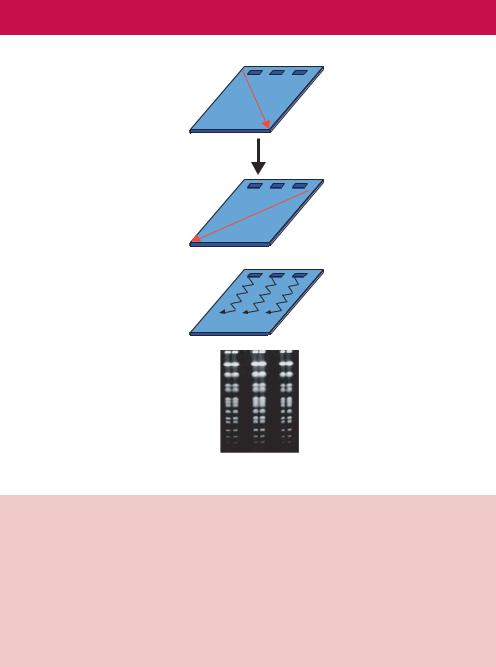
96 BASIC TECHNIQUES IN GENE ANALYSIS 2
(a) |
− |
|
|
Direction of DNA |
+ |
migration |
|
|
Switch current |
|
|
− |
Direction of DNA
migration
+
(b)
DNA zig-zags down the gel
(c)
Separation of yeast chromosomes by PFGE
Figure 2.17. Pulsed-field gel electrophoresis (PFGE). (a) The switching of the electric current during PFGE. Current is applied across the gel for a defined period – the pulse time – which is often in the range of 0.5–2 min. After this time the direction of the current is switched. (b) The repetitive switching of the current means that the DNA will zig-zag down the gel. The original PFGE technique used two non-homogenous electric fields to change the direction of DNA migration during electrophoresis (Schwartz and Cantor, 1984). The zig-zagging motion allowed the separation of large DNA molecules, but the nonhomogenous electric field resulted in bowed DNA banding patterns. (c) Using homogenous electric fields, straight-line separation patterns are obtained, like the separation of whole yeast chromosomes shown here
lane-to-lane comparisons. The simplest approach to obtaining straight lanes is termed field inversion gel electrophoresis (FIGE), which uses parallel electrodes to assure an homogeneous electric field. FIGE works by periodically inverting the polarity of the electrodes during electrophoresis. Because FIGE subjects the DNA to a 180◦ reorientation, the DNA spends a certain amount of time moving backwards. The 180◦ reorientation angle of FIGE results in

2.5 GEL ELECTROPHORESIS |
97 |
|
|
a separation range most useful under 2000 kbp. Furthermore, FIGE has mobility inversions, in which larger DNA can move ahead of smaller DNA during electrophoresis. The use of homogenous electric fields in conjunction with PFGE also results in the DNA running in a straight line. For example, contour-clamped homogeneous electric field (CHEF) electrophoresis reorients the DNA at smaller oblique angle, generally between 96 and 120◦. This causes DNA to always move forward in a zigzag pattern down the gel, but the DNA does not move from its lane and straight-line patterns are obtained (Figure 2.18).
Several parameters act together during PFGE to affect the effective separation range of a particular gel. These include the type and concentration of agarose used, the buffer composition, the buffer temperature, the electric field strength, the reorientation angle etc. However, the pulse time is primarily responsible for changes in the effective separation range. The pulse time is the duration of the each of the alternating electric fields. Shorter pulse times lead to separation of shorter DNA molecules because the smaller DNA fragments will begin to move more quickly upon reorientation than the larger fragments. Similarly, longer pulse times lead to separation of larger DNA molecules (Figure 2.19).
The advent of PFGE meant that single DNA molecules representing whole chromosomes could be separated with ease using gels. Large DNA molecules such as intact chromosomes are easily sheared and also difficult to pipette due to their high viscosity. The need for intact material during electrophoresis is obvious. Attempts to separate partially degraded or truncated material will result in the smearing of bands, and consequently gels become difficult to interpret. The solution to this problem is to first embed unbroken cells (e.g.
FIGE |
|
CHEF |
− |
A− |
B− |
|
+ |
B+ |
A+ |
|
Figure 2.18. The two main types of PFGE. Field inversion gel electrophoresis (FIGE) is where the current is switched back and forth at a 180◦ angle to the direction of overall DNA movement. Contour-clamped homogenous electric field (CHEF) electrophoresis systems employ multiple electrodes such that the precise position of the electric field can be accurately varied