

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf358 ENGINEERING PLANTS 1
infection by a second, related virus. The expression of the tobacco mosaic virus coat protein within plants, which is not sufficient to cause infection, does yield resistance to virus infection (Abel et al., 1986). Additionally, plants expressing RNA sequences complementary to the coat protein coding region and the 3 untranslated region were protected from infection (Powell et al., 1989). The mechanism of resistance to superinfection is not well understood, although the expression of the coat protein in different tissues of the plant will give rise to different levels of viral resistance (Clark et al., 1990). The expression of viral proteins in plant cells has also raised the possibility that other viruses, that are still able to infect the plant, may be able to ‘inherit’ the expressed coat protein. To date, there is no clear evidence that this type of problem might actually occur.
11.2.5Fungal Resistance
Plants have been engineered to have an increased resistance to various fungal infestations. This can be achieved by expressing antifungal proteins within the plant. For example, transgenic tobacco seedlings constitutively expressing a bean chitinase gene under control of the cauliflower mosaic virus 35S promoter showed an increased ability to survive in soil infested with the fungal pathogen Rhizoctonia solani and delayed development of disease symptoms (Broglie et al., 1991). Chitinases hydrolyse chitin, an unbranched polymer of β(1→4) linked N-acetyl-D-glucosamine (GlcNAc) that is found in the cell walls of many fungi. An alternative approach is to transfer sets of avirulence genes from the pathogen into the plant (Melchers and Stuiver, 2000).
11.2.6Terminator Technology
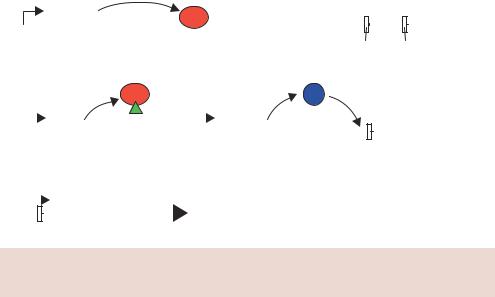
Perhaps the most controversial use of plant manipulation is the ability of producers to sell seeds that will produce plants for a single crop only. That is, seeds can be produced that will grow into normal plants, but the resulting seeds are sterile. This terminator technology (Figure 11.9) ensures that growers cannot save some of their own seeds for use in the following year, but must buy fresh seeds from the producer. The ability to grow sterile, but otherwise normal, seeds relies on the insertion of three genes into the plant:
•the gene encoding the tetracycline repressor (TETR) under the control of a constitutive promoter,
|
|
|
|
|
|
|
|
|
11.2 COMMERCIAL EXPLOITATION OF PLANT TRANSGENICS |
359 |
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
− Tetracycline (producer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
TETR |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
TetR |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Block |
|
RIP |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
tetO |
|
Cre |
|
|
|
Plea |
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
loxP loxP |
|
|
|
|
|
||||
|
|
+ Tetracycline (soak seeds before sale) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||
|
|
|
|
|
|
|
|
|
TETR |
|
|
Cre |
|
|
|
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
|
TET |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
TetR |
|
|
|
|
|
|
|
|
|
|
|
|
Cre |
|
|
|
|
|
|
|
RIP |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
tetO |
|
|
|
|
Plea |
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Excision of block |
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
− Tetracycline (farmer) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sterile seed |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
RIP |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
Plea |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
Figure 11.9. Terminator technology for the production of sterile seeds. See the text for details
•the gene encoding the Cre recombinase protein, whose expression can be inhibited by the binding of TETR to its promoter, and
•a gene that can be expressed late in embryogenesis that will make the seed sterile. As shown in Figure 11.9, the gene for a ribosome inactivating protein (RIP) is controlled from a promoter that is only active during late embryogenesis (lea; the promoter from the late embryogenesis abundant gene). RIPs are plant enzymes that are potent inactivators of eukaryotic protein synthesis (Stirpe et al., 1992). RIPs inactivate eukaryotic ribosomes by enzymatically cleaving a specific A residue in the 28S rRNA. This irreversible modification renders the ribosome unable to bind elongation factors and blocks translation.
The transgenic plant is constructed such that the RIP gene cannot be produced due to the presence of a blocking DNA sequence inserted into it. The blocker sequence is flanked by loxP recombination sites. The resulting plant is able to grow normally and produce normal, fertile seeds. The producer then soaks the seeds in tetracycline to inhibit the binding of the tetracycline repressor to the Cre gene, which can then be transcribed. The Cre protein catalyses the excision of the blocker DNA from the RIP gene. The seed is still capable of producing apparently normal plants, but the seeds it produces are sterile due to the expression of the RIP gene late in embryogenesis. The sterile seeds retain all the nutritional value of their normal counterparts, but are unable to form new plants.

360 ENGINEERING PLANTS 1
This immensely elegant way of protecting the investment made in transgenic crops was greeted with a massive public outcry when it was first patented in 1998 and 1999. The potential dangers of the terminator gene spreading from the plant to other organisms, coupled with the, real or perceived, control given to the seed producers, particularly in Third World countries, has meant that the technology has yet to be implemented.
11.3Ethics of Genetically Engineered Crops
A detailed discussion of the ethics of crop genetic engineering programmes is beyond the scope of this text. The engineering of plant traits has been occurring for thousands of years and the introduction of DNA technology has allowed, and will allow, many novel and important traits to be imparted. The safety of the resulting crops, both in terms of the edible product and potential effects on the environment, need to be rigorously assessed. The only realistic way that this can be achieved is through the careful design and thorough analysis of crop trials in a natural setting. Only then will any potentially harmful side-effects of the engineering process be identified.


362 ENGINEERING ANIMAL CELLS 21
for more than a few generations, a secondary cell line may be produced. Cell lines contain many cells that are essentially identical. Most cell lines will divide a relatively small number of times (10 –20) before entering senescence. Because these cells will die on their own, they are called mortal cells. Some cell lines, however, do not proceed to senescence and are described as immortal. These cells are said to be ‘transformed’ in that they have undergone a change to make them malignant or immortal. Examples of immortalized cell lines include
•HeLa cells (derived from a 31-year-old black woman named Henrietta Lacks, who died of cervical cancer in 1951) and
•Chinese hamster ovary (CHO) cells.
The changes that occur within these cells to make them immortal may result from a viral infection or other change within the cell that leads to unregulated cell division and growth. Here, the term transformation refers to this process rather than the uptake of foreign DNA as we have used it previously. In higher eukaryotes the process of inserting foreign DNA into cells is called transfection.
12.2Transfection of Animal Cells
The insertion of foreign DNA into animal cells is a two-stage process in which the DNA is firstly taken up by the cells (transfection), and secondly integrated into the genome. It is possible to generate stable animal cell lines that harbour extra-chromosomal vectors. For example, plasmids bearing the Epstein –Barr virus nuclear antigen (EBNA-1) and origin of replication (oriP) can be maintained within some primate and canine cell lines but not in rodent cell lines (Yates et al., 1985). However, most stable cell lines that ‘permanently’ contain the gene of interest depend upon the stable non-homologous integration of foreign DNA into the host chromosome. The transfection of cells is a far more efficient than integration of foreign DNA into the genome. Consequently, large numbers of cells can be transfected with foreign DNA, where it is maintained within the nucleus, but do not integrate it into their genome. The process of DNA uptake into animal cells may thus be described as being either transient or stable, depending how long the DNA is maintained within the cell.
•Transient transfection. If the foreign DNA transfected into cells is not integrated into the genome and does not contain an animal origin of replication, the cell will maintain it for a short period before it is either degraded or diluted from the cell during division. During the period in which the foreign DNA is inside the cell, it is treated just as any other DNA
12.2 TRANSFECTION OF ANIMAL CELLS |
363 |
|
|
sequence – genes contained within it may be expressed provided they are under the control of a suitable promoter, and the encoded protein produced. Transient transfection represents a rapid way to analyse foreign genes and gene products within cells. For example, many types of gene expression assay rely on the transfection of cells with an appropriate reporter gene, and the collection of data some 24 –48 h later. Relative to the generation time of most cell lines ( 16 –24 h), this represents a relatively short time between transfection and cell harvesting. In general, transient transfection protocols need to be optimized for each cell type being analysed due to inherent differences in DNA uptake efficiencies.
•Stable transfection. Rather than being maintained in an extra-chromosomal state within the nucleus of a host cell, the foreign DNA becomes integrated into an apparently random location within the genome. Once integrated, the foreign DNA is replicated together with the host chromosome in which it resides and gene expression can occur, providing that the foreign gene is controlled by suitable transcriptional control elements. Stable cell lines are required for the production of large amounts of a recombinant protein over a prolonged period of time. Technical difficulties in producing stable cell lines, arising mainly from the inefficiency of the integration process, mean that a clonal cell line can take several months to produce (Power and Meyer, 2000). This timescale often precludes their production.
The transfection of animal cells with genes from other organisms dates back over 40 years (Szybalska and Szybalski, 1962). In these experiments, the uptake of naked DNA into animal cells was an incredibly rare event. Animal cells are enclosed by a single membrane, called both the cell membrane and the plasma membrane. It is composed of a double layer of lipids containing a variety of proteins. The membrane is relatively impermeable and, under normal circumstances, allows only certain materials to pass into and out of the cell. In tissues, cells are also surrounded by extra-cellular matrix (ECM) – a mixture of proteins and proteoglycans – that are involved in cell –cell signalling, wound repair, cell adhesion and tissue function. Most cultured animal cells do not possess an ECM and are therefore relatively easy to insert DNA into compared with their native counterparts. As we have seen with other forms of transformation, the efficiency of the process can be greatly increased by various treatments, which are discussed below.
12.2.1Chemical Transfection
The efficiency of naked DNA transfection can be greatly improved if the DNA is precipitated in the presence of the cells. This can be achieved simply by
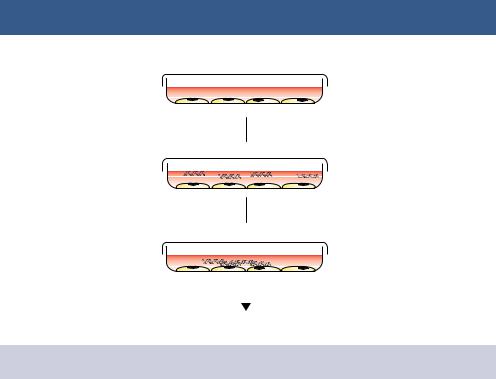
364 ENGINEERING ANIMAL CELLS 21
Cells growing in culture dish
Add DNA
 and CaCl2
and CaCl2
Allow precipitate
 to form slowly
to form slowly
Transfection
Figure 12.1. The transfection of animal cells using calcium phosphate precipitation
washing cultured cells in a phosphate buffer, adding the DNA, and then adding calcium chloride to the mixture (Figure 12.1). Under these circumstances, it is thought that the precipitate settles on the surface of cells and is then internalized through endocytosis (Orrantia and Chang, 1990). Some of the DNA will be released into the nucleus, where it may be expressed, or become integrated into the genome. Transfection efficiencies using calcium phosphate, as judged by the percentage of cells that take up the DNA, can be quite low, in the range of 1 –2 per cent (Graham and van der Eb, 1973), but can be increased if very high-purity DNA is used and the precipitate allowed to form slowly (Chen and Okayama, 1988).
12.2.2Electroporation
We have previously discussed the transformation of bacterial cells based on electroporation (Chapter 2). The technique also works well for animal cells (Wong and Neumann, 1982). By varying the electric field strength, and the length of time the cells are exposed to the electric field, it is possible to optimize electroporation parameters for almost any cell type.
12.2.3Liposome-mediated Transfection
Liposomes are spheres of lipid that can be used to transport molecules into cells. Classical liposomes are composed of a sphere of lipid bilayer surrounding

12.2 TRANSFECTION OF ANIMAL CELLS |
365 |
|
|
the molecule to be transported, and promote transport after fusing with the cell membrane. Cationic lipids (those bearing a positive charge) are used for the transport of nucleic acid. Cationic lipids naturally result in the formation of unilamellar liposome vesicles (Schaefer-Ridder, Wang and Hofschneider, 1982) that, rather than encapsulating the DNA within the liposome, are thought to bind along the surface of the negatively charged DNA to form clusters of aggregated vesicles over its length (Figure 12.2). Cationic liposomes
(a)
|
|
|
|
O |
Cl− |
|
|
|
|
|
O |
H |
|
|
|
|
|
|
|
N+ (CH |
) |
|
|
|
|
|
|
|
3 |
||
|
|
|
H |
H |
3 |
|
|
|
|
|
H |
|
|
||
|
|
|
|
H |
|
|
|
(b) |
+ |
+ |
+ |
|
|
|
|
|
|
|
|
|
|||
|
+ |
|
+ |
|
|
|
|
|
+ |
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
+ |
|
+ |
|
|
|
|
|
+ |
|
+ |
|
|
|
|
|
+ |
|
+ |
|
|
|
|
|
+ |
+ |
+ |
|
|
|
|
|
|
|
|
|
|
|
(c)
Liposome fusion with cell membrane and insertion of DNA into the cell
Figure 12.2. The introduction of DNA into cells using cationic liposomes. (a) The chemical structure of DOTMA (N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride), a cationic lipid used in the formation of liposomes (Felgner et al., 1987). (b) The structure of a cationic liposome. The positively charged ‘heads’ project outwards from a hydrophobic core formed by the lipid ‘tails’. (c) The surrounding of a DNA fragment by cationic liposomes prior to its insertion into the cell after fusion with the cell membrane

366 ENGINEERING ANIMAL CELLS 21
can be formed from a variety of cationic lipids, e.g. DOTAP (N-1(-(2,3- dioleoyloxy)propyl)-N,N,N-trimethylammoniumethyl sulphate) and DOTMA (N-(1-(2,3-dioleoyloxy)propyl)-N,N,N-trimethylammonium chloride). These are commercially available lipids that are sold as in vitro transfecting agents, with the latter sold as Lipofectin . Commercially available liposome formulations usually include a neutral lipid such as DOPE (dioleoylphosphatidylethanolamine) in order to facilitate liposome formation and cell membrane fusion. Cationic liposomes are able to interact with the negatively charged cell membrane more readily than uncharged liposomes, with the fusion between cationic liposome and the cell surface resulting in the delivery of the DNA directly across the plasma membrane (Figure 12.2). Additionally, material brought into the cell through cationic liposomes appears to be able to escape endosome mediated degradation that otherwise presents a major barrier to transfection via endocytosis. Liposome mediated DNA delivery is technically easy, highly reproducible, and very efficient. It is therefore often the method of choice for routine cell line DNA transfection.
12.2.4Peptides
A number of peptide sequences have been shown to be able to bind to, and condense, DNA to make it more amenable for entry into cells; e.g. the tetrapeptide serine –proline – lysine – lysine (located in the C-terminus of the histone H1 protein) promotes DNA transfection (Schwartz et al., 1999). The positively charged lysine amino acid side chains (see Appendix A) help to counteract the negatively changed phosphate DNA backbone and allow the DNA molecules to pack closely to each other. Rational design of peptide sequences has also been used to develop synthetic DNA binding peptides. For example, the peptide tyrosine –lysine –alanine –(lysine)8 –tryptophan –lysine has been shown to be very effective at forming complexes with DNA (Gottschalk et al., 1996). DNA binding peptides that can be coupled to cell-specific ligands can also be synthesized, thereby allowing receptor mediated targeting of the peptide/DNA complexes to specific cell types. One such approach has been to synthesise a cationic peptide based on 16 lysine residues and an arginine –glycine –aspartic acid (RGD) peptide sequence. The RGD sequence facilitates the binding of the peptide/DNA complex to an integrin receptor on Caco-2 cells in vitro (Harbottle et al., 1998). Synthetic peptides have also been developed that enhance the release of the peptide/DNA complexes from the endosome following endocytosis.
12.2.5Direct DNA Transfer
DNA may be physically injected directly into the nucleus of cultured cells (Capecchi, 1980). This approach is somewhat technically difficult and
12.3 VIRUSES AS VECTORS |
367 |
|
|
labour intensive in that cell nuclei must be injected with the foreign DNA on an individual basis, but is especially useful for large cells. For example, the oocytes of the African clawed frog Xenopus laevis, which can be harvested in large numbers from the ovaries of adult female frogs, are approximately 1 mm in diameter and have a correspondingly large nucleus. DNA, or RNA, injected into the nucleus is usually expressed in a mosaic pattern during the development of the frog (Melton, 1987). Integration of injected foreign DNA occurs at a very low frequency, but the expression of the foreign gene can be followed for many cell generations. The efficiency of the integration process varies considerably in different organisms. The direct injection of DNA into certain animal tissues, particularly into muscle and skin, has also been shown to produce high levels of transgene expression (Wolff et al., 1990). For example, patients suffering from the genetic skin condition lamellar ichthyosis (in which the skin is scaled and reddened), caused by a loss of transglutaminase 1 (TGase1) expression, show some skin regeneration following repeat injections of plasmid DNA encoding the TGase1 gene (Choate and Khavari, 1997). The restoration of TGase1 expression occurred in the correct location of the skin. Further analysis, however, revealed that the pattern of expression was non-uniform and failed to correct the underlying histological and functional abnormalities of the disease. Direct DNA injection has also been applied to the gene therapy (see below) of cancer, where the DNA can be injected either directly into the tumour or can be injected into muscle cells in order to express tumour antigens that might function as a cancer vaccine. Although direct injection of plasmid DNA has been shown to lead to gene expression, the overall level of expression is generally lower than that obtained with liposomes. We will return to direct DNA injection later when we look at the production of transgenic animals (Chapter 13).
12.3Viruses as Vectors
The relatively low efficiency of foreign DNA integration into animal cells, combined with the lack of naturally occurring extra-chromosomal plasmids, led to the manipulation of viruses as potential vectors for gene transfer.
12.3.1SV40
The first mammalian cell viral vector to be developed was based on the simian virus 40 (SV40) (Hamer and Leder, 1979). SV40 is a primate double-stranded DNA tumour virus whose genome is 5243 bp in size (Figure 12.3). Genes are