

John Wiley & Sons - 2004 - Analysis of Genes and Genomes
.pdf10.4 ANTISENSE AND RNA INTERFERENCE (RNAi) |
329 |
|
|
size or morphology (Giaever et al., 2002). Screening like this is a fairly crude measurement of the function of individual genes, but the analysis of different sets of genes required for growth under different conditions can be informative. Additionally, deletion analysis can be combined with gene expression profiling (microarrays, see above) to compare expression patterns between the wild-type and the mutant strains to give further clues to gene function. For example, the deletion of the TUP1 transcriptional repressor in yeast results in increased transcription of three per cent of yeast genes (DeRisi, Iyer and Brown, 1997). These genes are likely to be repressed the TUP1 protein in wild-type cells.
10.4Antisense and RNA Interference (RNAi)
The construction of gene knockouts in higher eukaryotic organisms is not as straightforward as that that described above for yeast, either as a result of low rates of homologous recombination, or the lack of suitable genetics. For some time, however, it has been known that the expression of antisense RNA (representing the sequence of the opposite sense to mRNA) can inhibit gene expression (Green, Pines and Inouye, 1986). Antisense sequences can be produced within cells by inverting the coding sequence of a gene with respect to its promoter such that the complementary strand is transcribed. The formation of a base-paired RNA duplex between the mRNA and the antisense RNA is thought to interfere with either RNA processing or translation, and the encoded protein is produced at much lower levels. For example, tomato plants expressing an antisense version of the polygalacturonase gene, the product of which is involved in softening and over-ripening, produce approximately five per cent of the normal polygalacturonase protein levels and have longer shelf-life and increased resistance to bruising (Smith et al., 1988). The expression of antisense RNA within cells, or the introduction of antisense oligonucleotides into cells, can have dramatic effects on the production of the protein encoded by the corresponding mRNA. Introducing an inducible antisense expression vector into cells may bring about conditional gene silencing. For example, the expression of antisense RNA from a tetracycline inducible promoter (see Chapter 8) may allow specific inhibition of protein production in the presence of tetracycline only (Handler and Iozzo, 2001). The efficiency of antisense inhibition can vary widely between species and individual genes, and specific antisense sequences may lead to a non-specific inhibition of protein production (Cohen, 1991).
It has also been discovered that the expression of individual genes in a variety of eukaryotes can be reduced dramatically through the introduction of specific double-stranded RNA molecules into cells. This phenomenon, termed

330 POST-GENOME ANALYSIS 10
RNA interference (RNAi), was first noticed during RNA injection experiments into the nematode Caenorhabditis elegans. The injection of either the sense or the antisense RNA strands of a particular gene into the organism caused little reduction in the expression of the gene. However, co-injection of both the sense and antisense RNA strands caused a massive reduction in the expression of the gene (Fire et al., 1998). A number of other experimental observations were made.
•The injection of double-stranded RNA for specific genes into C. elegans caused a specific disappearance of the corresponding gene product from both the somatic cells and the F1 progeny.
•Double-stranded RNA was able to inhibit gene function at a distance from the site of injection and appeared to be able to cross cell boundaries, suggesting that a small diffusible molecule may be responsible for the repressing effect.
•Only double-stranded RNA sequences from exons had any effect on protein production with sequences from introns having no effect.
•Relatively small double-stranded RNA sequences – considerably less than the full gene sequence – effectively turn off the production of the protein encoded by the corresponding mRNA.
•Very small amounts of double-stranded RNA, representing only a few molecules of double-stranded RNA per cell, were required to repress protein production, suggesting that a catalytic or amplification process was occurring.
RNAi can even be induced in C. elegans through the ingestion of doublestranded RNA. E. coli cells expressing specific double-stranded RNAs were fed to worms, and 40–80 per cent of the worms showed the specific phenotype associated with silencing (Timmons, Court and Fire, 2001). RNA silencing has been used extensively to analyse the function of genes in C. elegans. The ease with which silencing can be achieved and its overall effectiveness has made it a powerful tool in gene analysis. C. elegans has six chromosomes, and virtually all of the genes on two of these have been knocked out using RNAi (Gonczy¨ et al., 2000; Fraser et al., 2000). Knock-outs of a number of the genes result in sterile or embryonic lethal phenotypes that make additional analysis difficult, but many other genes can be assigned distinct functions based on this type of analysis (Kamath et al., 2003).
As with the introduction of antisense RNA, double-stranded RNA molecules can also be produced inside cells rather than being added to them. For

10.4 ANTISENSE AND RNA INTERFERENCE (RNAi) |
331 |
|
|
example, in trypanosomes, integrative plasmids have been constructed in which a trypanosome gene is under tetracycline (Tet) control and transcribes a head to head gene fragment with a spacer between the fragments to produce a double-stranded hair-pin RNA that induces RNAi (Shi et al., 2000). Initially, it was thought that RNAi might be limited in its scope, since the expression of double-stranded RNA in most mammalian cells results in a general downregulation of protein synthesis rather than a gene-specific effect (Hope, 2001). However, it was found subsequently that the expression of 21-nucleotide RNAs, called small inhibiting RNAs (siRNAs), paired so that they have a twonucleotide 3 -overlap, are able to down-regulate the expression of specific genes in mammalian cells (Elbashir et al., 2001). The repressive effects of RNAi in mammalian cells are not as great as that observed in flies or worms, but RNAi has great potential for determining gene function in a variety of previously genetically intractable organisms.
Box 10.1. The mechanism of RNAi
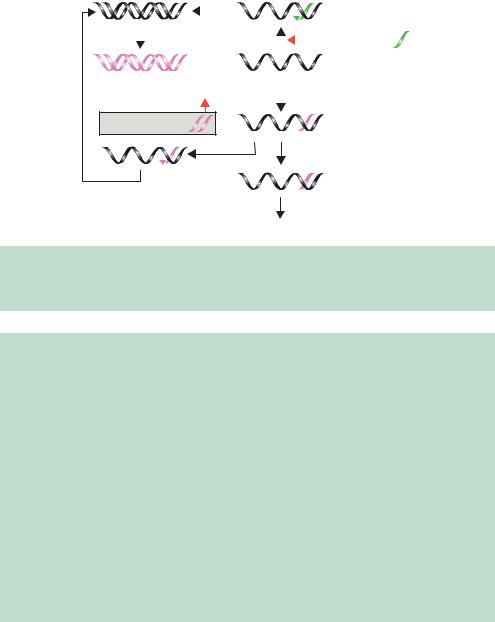
The molecular mechanism of RNA interference is not fully understood at present. RNAi has been shown to be a post-transcriptional phenomenon (Montgomery, Xu and Fire, 1998), but how does it work? The expression of double-stranded RNA induces the specific degradation of the mRNA to which it is complementary (Ngo et al., 1998). For example, in Drosophila, the expression of a 540-nucleotide double-stranded RNA corresponding to the cyclin E gene (a critical component of cell cycle control) induces cell cycle arrest and the production of 21–25-nucleotide RNA fragments that are homologous to the cyclin E gene – to both the sense and antisense sequences (Hammond et al., 2000). RNA degradation is ATP dependent, does not require the presence of the corresponding mRNA and is catalysed by a sequence-specific nuclease (Zamore et al., 2000). Through genome sequence comparison analysis, the Drosophila and C. elegans genomes are found to contain only three types of double-strand-specific RNase enzyme (Bernstein et al., 2001). One of these, named RNase III or Dicer, has been shown to be responsible for RNAi. The depletion of Dicer activity from cells results in the loss of the ability to silence genes by RNAi (Hutvagner´ et al., 2001).
The short interfering RNA molecules (siRNA) generated from doublestranded RNA serve as primers to transform the target mRNA into doublestranded RNA, which can then be degraded to generate new siRNAs (Lipardi, Wei and Paterson, 2001). An RNA-dependent RNA polymerase enzyme may play a role in amplifying the double-stranded RNA, but this has yet to be
332 |
POST-GENOME ANALYSIS 10 |
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Double-stranded RNA |
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
RdRp |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dicer - |
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
Addition of |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
Ribonuclease III |
|
|
|
antisense RNA |
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Target mRNA |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
siRNA |
|||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Addition of double stranded RNA
RNA cleavage
RdRp
RISC activation
RNA degradation
Figure 10.8. The mechanisms of antisense inhibition and RNA interference. The addition or expression of either antisense RNA (shown in green) or double-stranded RNA (pink) within cells can cause specific mRNA degradation. See Box 10.1 for details
shown experimentally. A model for the molecular mechanism of RNAi is shown in Figure 10.8 (Nishikura, 2001). The introduction of the antisense RNA sequence (shown in green) to a specific mRNA results in the formation of a double-stranded RNA hybrid between the two. The action of an RNAdependent RNA polymerase (RdRp) produces a larger double-stranded RNA molecule that is degraded into the 21–23-nucleotide fragments by the Dicer RNase. These siRNAs are also capable of binding to the specific mRNA from which they were originally derived. When bound to the mRNA, some of the siRNAs will serve as templates for the RdRp to produce more siRNA (Hutvagner´ and Zamore, 2002). The net result of this process is that the target mRNA is efficiently degraded, either through participation in siRNA production, or though the activation of an RNA induced silencing complex (RISC) that will degrade the mRNA–siRNA duplexes (Martinez et al., 2002).
The mechanism of RNAi is complex (see Box 10.1), but its natural role seems to have evolved to protect cells against transposons and viruses that may produce double-stranded RNA during their replication process. Of course, there are many examples of double-stranded RNA that occur naturally within cells, for example as part of the spliceosome. These do not, however, induce
10.5 GENOME-WIDE TWO-HYBRID SCREENS |
333 |
|
|
RNAi, perhaps because this double-stranded RNA is also associated with cellular proteins. In plants, RNA silencing may be used as defence mechanism against viral infections. Viruses containing RNA genomes are strong inducers of RNA silencing since double-stranded RNA is formed during replication. Additionally, RNAi may confer immunity against closely related viruses. As discussed above, a mobile silencing signal (possibly double-stranded RNA) can spread from cell to cell in the plant to provide viral immunity. Some viruses are thought to circumvent RNA silencing by spreading rapidly throughout the plant (Voinnet, 2001).
10.5Genome-wide Two-hybrid Screens
We have already discussed the use of two-hybrid screens to detect specific protein–protein interaction and to clone potential interacting partners from cDNA libraries (Chapter 6). To perform this type of analysis on a genomewide scale requires that every possible ‘bait’ (Figure 6.8) be tested against every potential ‘prey’ from an organism so that a complete protein–protein interaction map for the organism can be deduced. Moreover, if every gene within the genome has been identified through sequence analysis, the need to construct and screen a complex cDNA library is negated. A far more systematic approach is to clone and analyse individually each protein–coding gene within the genome. So, in the case of a two-hybrid screen, every potential protein–coding gene must be fused, in the correct reading frame, to the sequence encoding a transcriptional activation domain. In the case of yeast, approximately 6000 individual plasmids must be constructed so that all baits may be tested. This is by no means a trivial task. Cloning on this scale can be achieved, however, through the PCR amplification of the 6000 yeast genes from genomic DNA using a specific primer set for each gene (Figure 10.9). The primers were constructed such that each of the forward primers had a specific common 5 -tail of 20 nucleotides, and each of the reverse primers had a different common tail. The common tails could then serve as priming sites for a second round of PCR using a single primer set for all 6000 PCR products (Uetz et al., 2000). During the second round of PCR, each of the amplified products has a common 50 bp sequence added to their 5 -ends and a different common 50 bp sequence attached to their 3 -ends. The PCR products are then mixed with a linearized plasmid that contains these common sequences at its ends and transformed into yeast cells. Within the yeast, the common sequences in the PCR product undergo homologous recombination with the linear plasmid to reform a circular plasmid that can subsequently be replicated. This process results in the insertion of each PCR product, and encoded gene, into a yeast
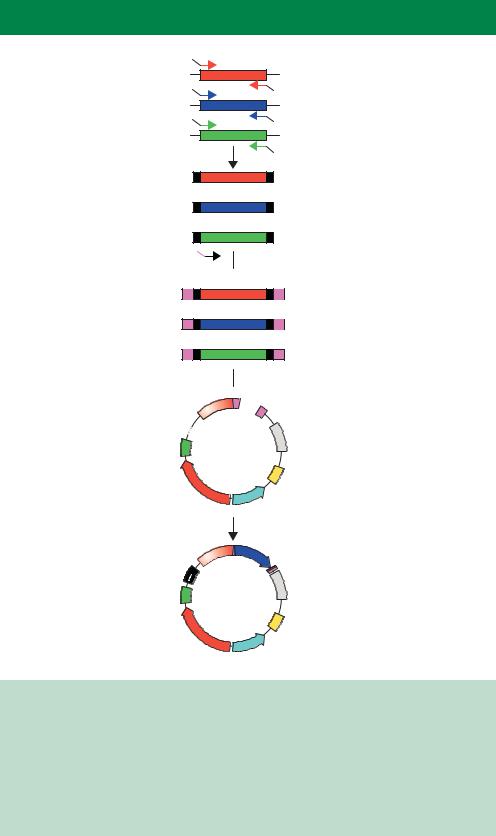


 common primers
common primers
 with linear vector
with linear vector

10.7 STRUCTURAL GENOMICS |
335 |
|
|
plasmid without the need for additional DNA manipulation. Similar ligationindependent cloning methods have also been devised for plasmid construction in E. coli cells (Donahue, Turczyk and Jarrell, 2002).
Once constructed, the 6000 yeast strains each producing a different transcriptional activation domain fusion (prey) can then be mated to a single yeast strain expressing a unique bait (fused to a DNA binding domain) to create a series of diploid yeast cells that all produce a single bait and each produce a different prey. The diploid cells can then be grown in small cultures in the wells of a micro-titre dish under conditions in which only an interaction between the bait and the prey will permit cell growth. Thus all of the potential interacting partners of a single bait can be identified in one experiment (Figure 10.10). The data obtained by repeating the screen using different baits can then be used to build up a comprehensive protein–protein interaction map (Uetz et al., 2000). To date, maps of this kind have only been produced for the protein interactions that occur inside yeast cells, but important information about the number of distinct protein complexes that exist within the cell has emerged. Many of the problems of two-hybrid screening that we discussed in Chapter 6 are also applicable here.
10.6Protein Detection Arrays
Rather than relying on the presence of an RNA transcript to infer the presence or absence of a protein within a particular cell, a better approach would be to detect the presence of the protein directly. Perhaps an even more stringent approach would be to detect the activity of individual proteins produced by a cell (Kodadek, 2002). The development of protein assays, akin to their DNA microarray counterparts, is still in its infancy. Some protein recognition chips are, however, available (Fung et al., 2001). These are composed of ligands, e.g. antibodies or small molecules, embedded in a surface such that they are immobilized, but still able to bind specifically to proteins. Cell extracts are then washed over the surface of the chip and bound proteins can be detected by mass spectrometry analysis.
10.7Structural Genomics
The importance of structural biology in advancing the understanding of molecular processes cannot be over-stated. The ability to visualize protein molecules in three dimensions at high resolution yields tremendous insights into their mechanism of action that could not have otherwise been obtained.

336 POST-GENOME ANALYSIS 10
Non-selective growth:
Selective growth:
Identify interacting partners
Figure 10.10. A genome-wide two-hybrid screen to identify protein–protein interactions. Yeast cells, each producing a single bait protein (in this case Pcf11p fused to the DNA binding domain of Gal4p), were mated with one of 6000 each producing a different prey (fused to the transcriptional activation domain of Gal4p) and grown in 384-well micro-titre plates. Yeast growth will only occur under selective conditions if an interaction between the bait and the prey occurs to activate the transcription of a reporter gene. The genes shown in the bottom panel are those prey fusions that potentially interact with Pcf11p. Images courtesy of Stan Fields (University of Washington), reprinted by permission from Nature (Uetz et al., 2000) copyright 2000 Macmillan Publishers Ltd
High-resolution structures are usually obtained using one of two methods – X- ray crystallography or nuclear magnetic resonance (NMR). In both cases, structure determination can be both time consuming and labourious. The first protein structures to be solved by X-ray crystallography were those of myoglobin and haemoglobin. Max Perutz began working on the structure of haemoglobin (molecular weight 67 kDa) in 1936 and finally solved the
10.7 STRUCTURAL GENOMICS |
337 |
|
|
structure in 1959 and published in 1960 (Perutz et al., 1960). These days, protein structure determination by X-ray crystallography is a great deal faster, primarily due to advances in computational power, but still relies on many of the techniques that Perutz and his colleagues pioneered. In general, both X-ray crystallography and NMR are dependent on the availability of large quantities of highly purified protein. Traditionally, protein structures have been solved on a piecemeal basis. Someone working on a biologically interesting protein finds that they are able to produce large quantities of it and then attempts structural analysis. The availability of genome sequences, however, provides an alternative route to solving protein structures – based solely on genomic DNA sequences (Figure 10.11). This approach is currently being attempted on a variety of completely sequenced organisms. Analysis of this type has only been made possible through the automation of almost all parts of the structure determination scheme shown in Figure 10.11. For example, 1376 of the predicted 1877 genes (73 per cent) of the thermophilic bacterium Thermotoga maritime have been cloned into an E. coli expression vector such that the produced protein bears a poly-histidine tag (Chapter 8). 542 of these clones were able to produce sufficient purified protein to attempt crystallization, and successful crystallization conditions were identified for 432 proteins, representing 23 per cent of the T. maritime proteome (Lesley et al., 2002). It is likely that not all of these crystals will yield protein structures, resulting in further attrition. The data above shows that the major stumbling block to successful structure determination is the availability of purified protein. Many proteins produced using general methodologies like this will be insoluble and therefore not amenable to structural analysis. Membrane bound proteins and others that might be deleterious to E. coli cell growth may be difficult to produce. An alternative approach is to make the proteins for structural analysis in vitro where cell related problems may be overcome. In vitro transcription/translation systems have been used for many years. In general they operate using plasmid DNA in which the gene to be expressed is cloned downstream of an RNA polymerase promoter binding site, e.g. T7 or SP6. The plasmid is then mixed with a recombinant form of the polymerase to produce RNA. The RNA is translated in vitro using cell lysates – derived from either E. coli or rabbit reticulocytes (Turner and Foster, 1998). Such systems are, however, limited in the amount of protein that can be produced and will not usually yield sufficient for structural analysis. Coupled transcription/translation systems have recently been developed, where the reaction occurs in a chamber separated from the substrates and energy components needed for a sustained reaction via a semi-permeable membrane. Transcription and translation can take place simultaneously in the reaction chamber, while inhibitory reaction by-products