

Глава 6
РАСШИФРОВКА СПЕКТРОВ ЯМР
Под термином «расшифровка спектра» обычно понимают различные процедуры, связанные с обработкой экспериментального спектра (выделение сигналов, определение значений спектральных параметров, отнесение сигналов к определенный ядрам и т. д.) и интерпретацией спектру (установление структуры исследуемого вещества). В гл. 6 будут рассмотрены методы расшифровки спектров ЯМР *Н (§ 1—5) и I3C—{‘Н} (§ 6—8), представляющие наибольший интерес для химика-органика. Для того чтобы полнее охватить комплекс методов расшифровки, будет изложен анализ спектров ЯМР ‘Н с использованием стационарного ЯМР, а анализ спектров ЯМР 13С — {!Н} — с использованием импульсной Фурье- спектроскопии.
§ 1. Предварительная обработка обзорных спектров ЯМР >н
Распространенная и весьма важная стадия анализа обзорного спектра — предварительная обработка, включающая выделение' сигналов и ^групп сигналов в спектре, приближенное измерение химических сдвигов, интегральных интенсивностей и форм мульти- плетов.
Калибровка спектра
Анализ спектра, начинают с установления шкалы химических сдвигов. Для этого необходимо определить масштаб и нуль шкалы.
Масштаб. Масштаб, или цена деления, выражается в м.д./мм шкалы (иногда в Гц/мм шкалы). Практически все современные спектрометры ЯМР используют калиброванные бланки, на которых масштаб шкалы устанавливается заранее. Обычно длина бланка L составляет 300—500 мм. Масштаб шкалы сдвигов определяется из соотношения
M=(SW)/L,
где SW — ширина спектра (в Гц или м.д.). Для обзорных спектров с SW=600—1000 Гц масштаб М составляет примерно 1—3 Гц/мм.
Нуль шкалы. В спектроскопии ЯМР ‘Н используется обычно ■6-шкала ТМС (или иногда t-шкала; гл. 3, § 2). Для установления нуля шкалы необходимо определить положение сигнала ТМС в спектре. Способы определения нуля шкалы зависят от типа ядерной стабилизации и способа эталонирования химического сдвига. При использовании внутренней стабилизации по ТМС область, близкая к нулю шкалы, обычно не регистрируется из-за биений основной и контрольной частот. В этом случае нуль шкалы устанавливается по значениям меток частоты. В тех случаях, когда исследуемый раствор не содержит ТМС, нуль шкалы устанавливают с помощью вторичных эталонов, используя данные о химических сдвигах этих эталонов в 6-шкале (табл. 3.1).
Точность калибровки спектра. Точность калибровки определяется типом стабилизации, типом эталонирования и некоторыми условиями записи спектра. Важнейшие источники ошибок перечислены ниже.
Случайные ошибки, обусловленные выбранным масштабом.
Систематические ошибки из-за использования линейной экстраполяции шкалы.
Дрейф (для внешней стабилизации и других недостаточно эффективных систем стабилизации). Дрейф резонансных условий приводит к искажению масштаба шкалы, что обнаруживается при повторной (или многократной) записи спектров.
Ошибки в результате использования других эталонов. К такой методике следует относиться с известной осторожностью, поскольку вторичные эталоны могут испытывать влияние специфических межмолекулярных взаимодействий.
Сигналы и группы сигналов
%
В общем случае спектр ЯМР 'Н может быть представлен в виде суперпозиции отдельных сигналов, имеющих форму, близкую к лоренцовой. Однако на практике разложение спектра на отдельные сигналы или линии вызывает значительные трудности. Рассмотрим этот вопрос несколько подробнее.
Прежде всего в стационарном ЯМР идеальная лоренцова форма может быть возмущена из-за эффектов быстрого прохождения. Возможны и другие аппаратурные искажения, в частности неправильная установка фазы сигнала, влияние неоднородности магнитного'поля, наличие боковых сигналов от вращения.
Принципиально важным источником возмущения формы линии являются процессы химического обмена (гл. 4). В области промежуточных скоростей обмена контур линии не может быть представлен в виде суперпозиции лоренцовых форм, и требуется более точный анализ с помощью уравнений Блоха—Мак-К.оннела.
Наконец, достаточно общим фактором, определяющим контур экспериментального спектра, является перекрывание близких сигналов. При этом линии идеальной лоренцовой формы могут образовывать контур сложной формы. Разложение такого контура на отдельные линии лоренцовой формы может представить серьезные трудности.
Во многих случаях на стадии предварительной обработки достаточно разбить спектр ЯМР !Н на группы сигналов. Группой сигналов называют один или несколько сигналов (т. е. мультипле- тов), отделенных от других групп сигналов промежутком, не содержащим каких-либо сигналов. Отдельные компоненты группы сигналов могут быть обусловлены сигналами химически неэквивалентных протонов. Другая причина возникновения мультипле- та — спин-спиновые расщепления. Для спиновых систем, не содерт жащих ядер 19F, 31Р (или не обогащенных по изотопу 13С), выделение групп сигналов позволяет провести приближенное разделение протонов образца на отдельные группы.
При осмотре спектра следует вначале идентифицировать сигнал ТМС (обычно крайний справа, т. е. в сильных полях), сигналы растворителя и известных компонент раствора. Сигналы растворителей идентифицируют по значению химического сдвига (гл. 3, табл. 3.1) или по форме соответствующих мультиплетов. Следует идентифицировать также известные компоненты раствора (добавки GF3COOH, Н20, шифт-реагенты).
Интегрирование
Обзорный спектр, как правило, дополняют записью интегральной функции
V
I(vH) = A j g(v)dv, (6.1)
VK
где vK — нижний предел интегрирования, представляющий собой верхнюю границу резонансных частот (как правило, интегральную функцию записывают слева направо, т. е. в сторону уменьшения резонансных частот). Если нижняя и верхняя границы диапазона интегрирования выбираются так, чтобы весь исследуемый спектр оказался внутри диапазона, то соответствующее значение интеграла называется полным.
Для. индивидуальных соединений вводится понятие протонной единицы (п.е.)
1п.е==1полн/N, (6.2)
где N — число протонов в соединении. При этом физический смысл приобретает доля интеграла
V>
Pi — f 8 (v) dv/1 ПОЛИ) (6.3)
где область интегрирования ограничена диапазоном резонансных частот i-той группы сигналов. Для смеси m компонент с мольными долями pi и числами протонов Ni п.е. интеграла определяется так:
In.с. —*1полн/Л^ — 1пол„/£ PiN(. (6.4)
/=1
Ошибки интегрирования. Следует различать систематические и случайные ошибки интегрирования. Наиболее распространены систематические ошибки следующих типов: а) перекрывание сигналов, что вызывает ошибки в определении доли интеграла;
б) дрейф «нуля» интеграла, что приводит к характерным, накло- •нам кривой 1(у)(гл. 5, рис. 5.16,3); в дрейф резонансных уело* вий, что сопровождается смещением кривой I(v) отндсительно спектра g(v).
Случайные ошибки являются следствием нестабильности Электронного интегратора и шумов приемных схем. Величина случайной ошибки, приведенная к стандартной концентрации раствора, является важной характеристикой спектрометра. Абсолютная величина ошибки (среднеквадратичное отклонение или дисперсия) интегрирования cti может быть определена с помощью статистической обработки серии измерений.
Отношение интегральных интенсивностей двух сигналов представляет интерес для количественного анализа, например при исследовании констант равновесия форм А и В:
СПЕНТРОСНОПИЯ ЯМР 1
. дН% т л г. 8
М, 31
/z==itmh 44
ух, 63
= С^к' 115
. (j). — 0^н‘ 115
HJ_ II 144
l/L 197
JU 202
2 (-^-) Лр>=vj—<6-21> 245
откуда видно, что относительная ошибка имеет минумум при К=\.
Определение приближенных значений химического сдвига
В большинстве случаев группа сигналов может быть охарактеризована одним значением химического сдвига в 6-шкале. Для симметричных групп сигналов химический сдвиг определяется по положению центра симметрии. Для несимметричных мультипле- тов приближенное значение химического сдвига соответствует положению «центра тяжести» мультиплета. Центр тяжести группы сигналов находят с помощью интегральной кривой.
Рассмотрим процедуру предварительной обработки типичного обзорного спектра ЯМР ’Н (рис. 6.1, там же дана интегральная кривая). Об образце и спектрометре известно следующее.
Образец. Раствор в CCU органического соединения неизвестной структуры (~50 мг в 0,5 мл раствора). К раствору добавлен ТМС.
Спектрометр. Varian HA-100D, частота ЯМР 'Н — 100 МГц; непрерывный режим, внутренняя стабилизация по сигналу ТМС, ширина развертки 1000 Гц.
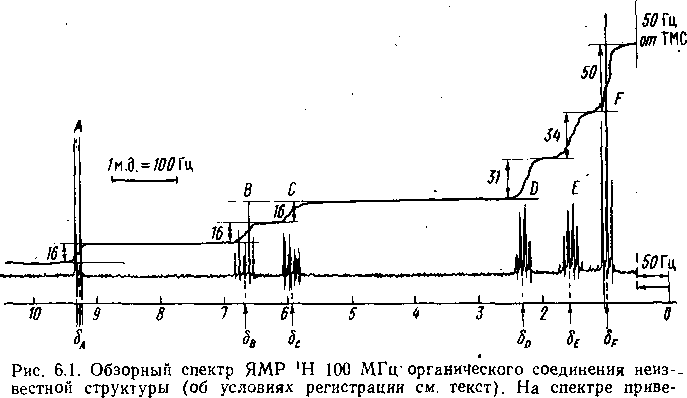
дена б-шкала химических сдвигов, масштаб (м. д.), метка частоты 50 Гц от ТМС. Интегральная кривая разбита на «ступени», приведена ведичииа «ступени» в мм «/-координаты. Вертикальными стрелками обозначены приближенные положения химических сдвигов групп сигналов А—F
Спектр. В правой части спектра оператором дана метка частоты: 50 Гц от ТМС. Согласно принятым критериям (гл. 5, § 2) качество спектра удовлетворительно.
"Калибровка шкалы. Для спектрометра Varian HA-100D ширина спектра 1000 Гц соответствует масштабу 2 Гц/мм или 1 м. Д./50 мм (длина калиброванного бланка L равна 500 мм). На рис. 6.1 приведен масштабный• отрезок, равный 1 м. д. Используя значение метки частоты 50 Гц и цену деления, определяют положение нуля шкалы (6=0,0 м. д.). На прямой, параллельной базисной линни спектра, проводят разметку шкалы (через 1 м. д.). Изучаемый спектр находится в пределах от 0,5 до 10,8 м. д. Поскольку спектр записан с использованием внутренней стабилизации по сигналу ТМС, можно пренебречь дрейфом и неточностью в определении нуля б-шкалы. Случайные ошибки в измерении химических сдвигов и констант спин-спииового взаимодействия могут быть обусловлены мелким масштабом спектра и достигать ±2 Гц. Систематические ошибки могут быть вызваны линейной экстраполяцией шкалы в область слабых полей, однако эти ошибки невозможно оценить из одного эксперимента.
Выделение групп сигналов. В спектре (рис. 6.1) легко идентифицируются шесть групп сигналов, отмеченных буквами A—F. Сигнал ТМС в спектре отсутствует; раствор не содержит дополнительных протонсодержащих компонент. Следовательно, все шесть групп сигналов А—F должны относиться к исследуемому веществу.
Интегрирование. Наиболее четкие «плато» интеграла обнаруживаются в следующих областях спектра: 1) до сигнала А; 2) между сигналами А я В; 3) между сигналами С и D«B остальных случаях имеется больший или меньший наклон «плато». В области за сигналом F наблюдается сильное «сползание» интеграла.
Определение химических сдвигов групп сигналов. Химические сдвиги сигналов Л—F находят по положениям приближенных центров симметрии этих сигналов.
Результаты предварительного анализа спектра, приведенного на рис. 6.1, суммированы в табл. 6^1.
Таблица
6.1
Результаты
предварительной обработки
экспериментального спектра ЯМР 1Н*
Группа
сигналов
*
Доля
интеграла
Химический
сденг м. д. (6-шкала)
А
0,094
9,30
В
0,094
6,65
С
0,094
5,90
D
0,193
2,30
Е
0,212
1,55
F
0,312
0,95
*
Спектр приведен
на
рис.
6.1.