

§ 1. Основные понятия динамической стереохимии
Согласно приближению Борна — Оппенгеймера движения ядер и электронов молекулы могут быть описаны раздельно. В другой формулировке это означает, что строение электронных оболочек может быть рассмотрено как функция координат ядер и, следовательно, электронные свойства могут быть получены на основании известной- структуры ядер молекулы, т. е. геометрии молекулы.
Положение ядер молекулы в трехмерном 'Пространстве можно описать с помощью потенциальной поверхности V(гь г2, ..., />), где г\, г2, ..., гх — координаты ядер в молекуле. Каждому набору значений координат гь г2, ..., Гу соответствует тот или иной потенциал V(r), причем устойчивым конфигурациям ядер будут соответствовать минимумы потенциала. Таким образом, задача нахождения геометрической структуры молекулы сводится к изу-' чению свойств потенциальной поверхности V(r\, г2, ..., rN).
Системы с одним минимумом потенциальной энергии
Допустим, что потенциальная поверхность V (г\, г2, ..., гЛ-) характеризуется единственным минимумом Кмин- При наличии большого числа частиц-довольно трудно наглядно представить поверхность V (гь г2, ..., rN), поэтому мы упростим рассмотрение анализом сечений поверхности вдоль некоторой эффективной координаты, проходящей через УШш- Сечение потенциальной поверхности вдоль координаты г имеет вид, приведенный на рис. 4.1.
Молекулы, имеющие единственный минимум на потенциальной поверхности, называются жесткими. Примером такой молекулы является метан СН4; протоны этой молекулы располагаются в вершинах правильного тетраэдра. Если в силу каких-то случайных воздействий координаты одного из атомов жесткой молекулы изменяются, то потенциал V(r) возвращает этот атом в исходное положение.
Однако атомы и в жестких молекулах не находятся в абсолютном покое даже при абсолютном нуле температур. Во-первых, молекулы могут двигаться поступательно как целое, что определяет процессы текучести и поступательной диффузии. Во-вторых, молекула может вращаться, что определяет процесс вращательной диффузии. Наконец, относительные положения ядер могут изменяться в результате колебаний около положения равновесия. Поскольку процессы, связанные с движением молекулы как целого, нас пока не интересуют, ограничимся рассмотрением колебательного движения.
m
Рис.
4.1. Потенциальная функция V(г)
молекулы. Минимум потенциальной
поверхности соответствует равновесному
расстоянию Гравн- Пунктирная линия
соответствует потенциалу вида (4.1)
с энергетическими уровнями гармонического
осциллятора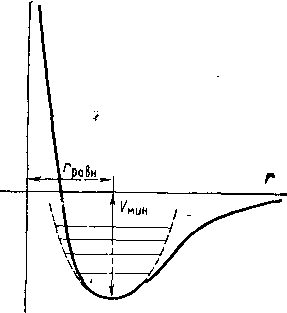
Гк) = Уммн- В результате решения уравнений для квантового осциллятора получают систему стационарных колебательных состояний, характеризующихся определенным квантовым колебательным числом и определенной энергией (рис. 4.1).
Энергии колебательных состояний существенно зависят от формы потенциальной поверхности. Известно, что вблйзи от минимума (т. е. около Гравн) форма потенциала V{г) может быть аппроксимирована параболой
V(r)toa?z, (4.1)
где г—г—грав„, коэффициент а представляет собой кривизну V(г). Квантовомеханические расчеты показывают, что колебательные уровни сближаются с уменьшением кривизны. Кривизна потенциальной поверхности представляет собой довольно важную характеристику жесткого состояния.
Молекулы с колебаниями малой амплитуды. Колебательный процесс представляет собой периодические движения, ядер около положения равновесия. Это движение можно охарактеризовать амплитудой колебаний I. Амплитуда колебаний определяется кривизной V(r); для таких жестких систем, как метан, бензол, норборнан и т. д., амплитуда колебаний составляет ~0,05А. Такие амплитуды колебаний можно считать малыми, однако следует иметь в виду, что даже в этом случае понятие структуры при
обретает вероятностный смысл. Структуру с колеблющимися ядрами можно описать с помощью функции плотности вероятности межатомных расстояний Р(г). Величина Р(г) представляет собой вероятность того, что межатомное расстояние находится в интервале от г до г-{-dr. Функция плотности вероятности Р(г) для жест-
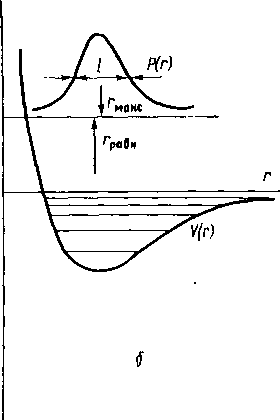
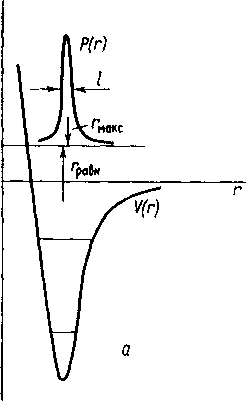 Рис.
4.2. Потенциальные функции У (г) для молекул
двух типов: а
— жесткие молекулы, характеризующиеся
глубокой потенциальной ямой и узкой
функцией распределения Р{г)\
б
— колебательно-нежесткие молекулы
с уплощенной ямой и колебаниями большой
амплитуды
Рис.
4.2. Потенциальные функции У (г) для молекул
двух типов: а
— жесткие молекулы, характеризующиеся
глубокой потенциальной ямой и узкой
функцией распределения Р{г)\
б
— колебательно-нежесткие молекулы
с уплощенной ямой и колебаниями большой
амплитуды
ких молекул с одним минимумом V(г) представляет собой колоколообразную функцию с максимумом при г=гмакс. соответствующим наиболее веройтному значению расстояния (рис. 4.2).
Поскольку потенциал V(r), как травило, несимметричен, значение Гмакс не совпадает с гРавН; обычно Г макс ^>Г рашь ТЭК K3K ОТ- талкивающая часть потенциала растет более резко.
Функция плотности вероятности Р(г) может быть введена для каждого колебательного состояния. Для возбужденных состояний функции Pi(r) аналогичны Л>(г), но из-за асимметрии потенциала обычно расстояние гМакс, наиболее вероятное для /-того состояния, возрастает с ростом квантового колебательного числа.
Для молекул с колебаниями малой амплитуды при комнатных температурах, как правило, занято только низшее (основное) колебательное состояние. Степень занятости возбужденных состояний pi можно оценит^ с помощью распределения Больцмана:
Pi=exp(—Ei/RT). (4.2)
Здесь Ei — энергия t-того уровня. Так как Ei = hiо, где о — фундаментальная частота колебаний, то для значений частот ш =
= 1000 см-1 и для комнатных температур получим величину р\ для 'Первого возбужденного состояния, равную ~ 1 %.
В спектроскопии ЯМР колебательные поправки могут оказаться важными при анализе двух экспериментальных фактов.
Химический сдвиг ядер, входящих в жесткие молекулы, зависит от температуры. Химический сдвиг ядра определяется структурой молекулы и, следовательно, зависит от типа колебательного состояния. В общем случае наблюдаемый сдвиг определяется усреднением по колебательным состояниям:
б = £ РД, (4.3)
/=0
где Pi — доли состояний, которые определяются согласно (4.2). Поскольку доли состояний зависят от температуры, то и наблюдаемый химический сдвиг оказывается зависящим от температуры. Экспериментальные исследования некоторых жестких моле- v кул показали, что «температурные коэффициенты» дб/дТ для химических сдвигов составляют ~ 10~4 м. д./°С для сдвигов ‘Н и ~ Ю~2 м. д./°С для сдвигов 13С. Эти -поправки невелики, однако три изучении спектров в широком температурном диапазоне (около 100°) они могут заметно превышать ошибки воспроизводимости. Необходимость учета колебательных поправок существенно усложняет изучение кинетики быстрых процессов с помощью ЯМР (гл. 4, § 2).
Химический сдвиг ядра зависит от изотопного состава молекулы. Экспериментальные данные показывают, что замена одного изотопа на другой в жесткой молекуле приводит к изменению химических сдвигов соседних ядер. Так, если в углеводородной цепи происходит Н—D замещение
Н—С*—Ср—Cv D—Са—Ср—Cv,
то наблюдается смещение резонанса 13С в сильные -поля: примерно на 0,3 м. д. для Са; примерно на 0,1 м. д. для Ср; примерно на 0,03 м. д. для Cv- При замене ядра 12С на 13С резонанс ближайшего протона смещается в сильные поля примерно на 0,002 м. д.:
Н—С12 Н—13С
Изотопные эффекты представляют самостоятельный интерес в спектроскопии 'ЯМР как метод расшифровки сложных спектров (гл. 6). Распространенным подходом для анализа изотопических эффектов является рассмотрение' колебательных поправок. В самом деле, при замене Н на D происходит уменьшение энергий колебательных уровней. Поскольку форма потенциальной поверхности при этом не изменяется, понижение энергий уровня приво-
дит к росту заселенностей возбужденных колебательных состояний, что и вызывает изменение химических сдвигов ядер молекулы в соответствии-с формулой (4.3).
Молекулы с колебаниями большой амплитуды. В этом случае потенциальная поверхность V(г) вдоль какой-либо из координат характеризуется малой крутизной (такие поверхности иногда называют уплощенными ямами). Уплощенные ямы характеризуются достаточной заселенностью возбужденных колебательных состояний (рис. 4.2, б), функция плотности вероятности Р (г) имеет значительно большую ширину, и амплитуды колебаний могут достигать значений околоТА, сравнимых с межатомными расстояниями в молекулах.
Типичным примером таких систем является енольная форма ацетилацетона (R = CH3)
н
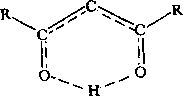
Полагают, что сечение потенциальной поверхности вдоль эффективной координаты, связывающей ядра кислорода, представляет собой уплощенную яму с малой кривизной. Протон между атомами кислорода характеризуется «размазанной» функцией распределения Р(г) и как бы «делокализован» в пространстве между двумя кислородами. Характерно, что химические сдвиги протона в связи О—Н...О характеризуются большим температурным коэффициентом: ~ 10~2 м. д./°С (это значение на два рорядка превышает типичные температурные коэффициенты для жестких молекул). Колебательная нежесткость, вероятно, свойственна всем системам с внутримолекулярными водородными связями ОН...О, МН...О, NH...N и др.
Таким образом, для молекул с колебаниями большой амплитуды понятие структуры как устойчивой трехмерной конфигурации утрачивается. Молекула приобретает элементы нестабильности. Свойства таких соединений сильно зависят от температуры и от среды.
Системы с двумя минимумами потенциальной
энергии. Динамические молекулы
В общем случае потенциальная поверхность V(г) может иметь несколько минимумов. Если барьер, разделяющий эти минимумы, сравнительно невелик, то в результате активации молекулярная система может обратимо переходить из одного минимума в другой.
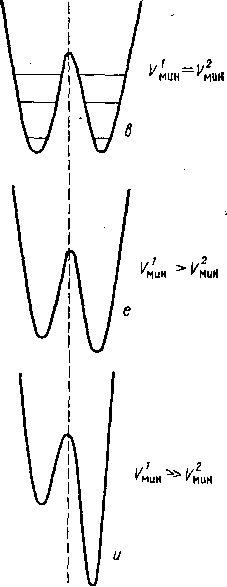
Рассмотрим для простоты случай двух локальных минимумов V-teaa и К2м№. Этим минимумам соответствуют структуры- Ф[ и ф2 и значения эффективной координаты реакции г\ и г2. Сечение типичной потенциальной поверхности этого типа обнаруживает
Рис.
4,3. Потенциальные поверхности для систем
с двумя мийимумами; а—в — вырожденные
системы; г—е—невырожденные
системы с состояниями сравнимо»
энергии; ж—и
— невырожденные системы с сильно
смещенными равновесиями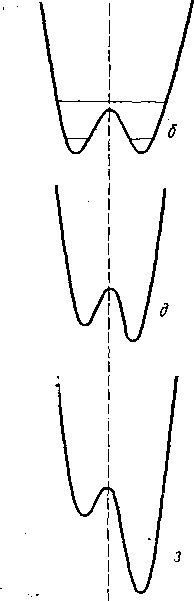
 максимум
V(r)
= V$
при значении
re(riCrQCr2).
При г<г;
и гг2
потенциальная энергия V
(г)
неограниченно возрастает, так что в
целом система находится в потенциальной
ямс. Все разнообразие форм потенциальных
поверхностей описывается двумя
■«переменными величинами: разностью
энергий b.V—V2—и
ве- f
личиной барьера Уб
(рис. 4.3).
максимум
V(r)
= V$
при значении
re(riCrQCr2).
При г<г;
и гг2
потенциальная энергия V
(г)
неограниченно возрастает, так что в
целом система находится в потенциальной
ямс. Все разнообразие форм потенциальных
поверхностей описывается двумя
■«переменными величинами: разностью
энергий b.V—V2—и
ве- f
личиной барьера Уб
(рис. 4.3).
Вырожденные системы. Прежде всего рассмотрим важный частный случай поверхностей, симметричных относительно г§. Такие поверхности характеризуются АУ—0. Формы Ф[ и Ф2 идентичны
и различаются только перестановкой идентичных ядер. Сиия- указанного типа называются вырожденными.
В общем случае обратимый процесс Ф^Фг можно охарактеризовать константой скорости k или временем жизни состояния т, причем
Л=1/т. (4.4)
В теории активированного комплекса зависимость константы скорости от температуры описывается с помощью уравнения
k т
k = x——ехр (— AG^/RT), (4.5)
h
f
где к — трансмиссионный коэффициент, учитывающий вероятность того, что система, достигая переходного состояния, перей- лет в альтернативную форму; къ — постоянная Больцмана; AG^ — свободная энергия активации
AG* = AH*—TAS*\ (4.6)
АНф и AS* — энтальпия и энтропия активации.
Учитывая слабую зависимость предэкспоненциального множителя от температуры, часто используют уравнение Аррениуса
6=Лехр(—EJRT), (4.7)
где А — частотный фактор; Еа — энергия активации. Сопоставление уравнений (4.5) и (4.7) дает
АН* = Еа — RT, (4.8а)
AG* = Ea-RT-TAS*. (4.86)
А — е • ехр (AS^/R). (4.8в)
Для многих типов процессов можно положить Л5=^=0 и х=1; тогда пренебрегая зависимостью частотного фактора от температуры, получим
k = 6,2 ■ 1012 ■ ехр (— V6/RT), (4.9)
где принимается Айф — АНф=Уб.
В табл. 4.1. приведены значения констант скоростей, рассчитанные с помощью уравнения (4.9) для трех температур: 200, 300 и 400 К для барьеров в диапазоне от 3 до 30 ккал/моль.
Системы, обладающие барьером ниже 5 ккал/моль, характеризуются высокими скоростями обмена во всем интервале температур. Эти системы, по существу, могут рассматриваться с помощью потенциальных поверхностей с одним (но уплощенным) минимумом, что позволяет отнести их к молекулам с большими амплитудами колебаний. С другой стороны, если барьер V§ превышает 25 ккал/моль, то обмен является медленным во всем тем- , лературном диапазоне. Структуры Ф1 н Ф2 в эюм случае соответствуют двум достаточно устойчивым формам соединения. Наконец, если барьер Уб лежит в диапазоне 5—25 ккал/моль, то скорости обмена имеют промежуточный характер. Системы такого типа называют динамическими или стереохимическими нежесткими.
Для дальнейшего изложения весьма важно рассмотреть вопрос о возможности экспериментального измерения констант скоростей. Любой метод измерения скорости химической реакции мож-
Таблица
4.1
Константа
скорости вырожденного обмена при
различных барьерах и температурах 200
, 300 и 400 К
Барьер,
Константа
скорости, Гц прн температуре
гкал/моль
кДж/моль
200
300
400
3
12,57
3,4
109
4-10'°
1,4
1011
5
20,95
2,3-107
1,4-109
1,1
1010
10
41,90
8,6-10
3-105
2,3
107
15
62,85
3,2-10-4
4,7-10
4,4
104
20
83,80
1,1-Ю-9
1,5-10~2
8,6
10
25
104,75
4,4-10-15
з,з-ю-6
1,6
ю-1
30
125,70
1,6-
ю-20
7,2-
Ю^10
3,2-
10-4
но охарактеризовать некоторой постоянной времени тм, учитыва- ющей «инерционные» свойства регистрирующего устройства. Вообще говоря, для данного регистрирующего метода постоянная времени лежит в некотором диапазоне значений ты, поэтому говорят о временной шкале метода. Из общих соображений очевидно, что метод, характеризующийся определенным значением тм (точнее, определенной временной шкалой), наиболее эффективно •применим для тех реакций, 'период полупревращения которых близок к тм. Если т<тм, то регистрирующее устройство не успевает следить за 'процессом. Напротив, если тЗ>тм, то динамическая система воспринимается как статическая.
Многие методы, используемые химиком-органиком, ,по существу относятся к числу визуальных методов. Эти методы не применимы для изучения сравнительно быстрых 'процессов, период полупревращения которых меньше 1 с.
Оптические методы (ИК, УФ) и методы дифракционного анализа относятся к числу методов с очень короткой постоянной времени, лежащей в диапазоне 10~12-М013 с. Подобные «быстрые» методы воспринимают динамическую систему как статическую. Если процесс обратим (Ф^Фг) и система находится в динамическом равновесии, .то отклик регистрирующего устройства не зависит от времени жизни.
Анализ большинства методов, традиционно используемых в химической кинетике, показывает, что важная группа процессов, идущих со скоростями 10—106 Гц, оказывается малодоступной для экспериментального исследования. Уникальное значение в связи с этим имеет использование метода ЯМР. Как будет показано ниже (§ 2), этот метод обладает временной шкалой, лежащей в диапазоне от 10-1 до 10~6 с.
Невырожденные системы. Формы Фг и Ф2 имеют различные энергии. Для описания невырожденных систем вводят константу равновесия /С, которая определяется как отношение содержаний двух форм:
# = _ ехр (— AVIRT), (4,10)
[4>il
где AV—Vz—Vi. Очевидно, что для вырожденных систем константа равновесия равна единице.
Потенциальные поверхности невырожденных систем с двумя максимумами могут быть качественно разделены на два класса. К первому классу следует отнести системы, характеризующиеся не слишком различающимися значениями Vi и У2; допустим для определенности, что |Vi—V2|<;2 ккал/моль. Такие системы, которые будем называть слабосмещенными равновесиями (рис. 4.3, г—е), показывают заметное содержание обеих форм; константа равновесия для этих систем не превышает 100. К другому классу систем относятся -потенциальные поверхности с резко различающимися значениями Vi и У2; допустим, | V2—V1\^>2 ккал/моль. Следует отметить, что moi существу такие сильносмещенные равновесия приближаются к жестким системам, поскольку наличие второго высоколежащего локального минимума можно рассматривать как несущественную деталь потенцналыюй поверхности.
Важно отметить, что невырожденные процессы включают не одну, а две химические реакции: ф,—)-ф2 и Ф2—►Фь Эти реакции характеризуются различным барьером активации, причем
Vg (12) = l^c(21) + А.17. (4.11)
Таким образом, скорости реакции в различных направлениях различны,причем
-*S-=-IL =/(, (4.12)
hi т2
где ть %2 — времена жизни в состояниях 1 и 2.
Для того чтобы единым образом описать скорости вырожденных и невырожденных процессов, вводят эффективное время жизни
х=р2Т1 = /?1Т2, (4.13)
где р\ — \—Р2■ Содержания форм р\ и /?2 могут быть выражены через константу равновесия
pi~KI
(1+^0.
р2=\/(\+К).
(4.14)
(4.15)
Для вырожденных случаев pi = /?2=l/2, t=ti/2=t2/2.
Для слабосмещенных равновесий энергии форм несильно различаются между собой, поэтому классификация потенциальных поверхностей, предложенная выше для вырожденных систем, применима в целом и для невырожденных систем. Так, если Т/б(12) й Уб(21) менее 5 ккал/моль, то соответствующая потенциальная (поверхность (рис. 4. 3, г) может быть рассмотрена как система с одним несимметричным уплощенным минимумом. В том случае, если Уб(21) и V(j(l2) больше 25 ккал/моль, формы Ф[ и Ф2 можно считать энергетически неэквивалентными жесткими формами соединения. В химии такие структуры традиционно называют изомерами. Наконец, наибольший экспериментальный интерес имеет случай барьеров, лежащих в диапазоне от 5 до 25 ккал/моль (рис. 4.3, д). В этом случае химически неэквивалентные формы соединения, называемые конформе^ами или ротамерамн, быстро превращаются друг в друга.
Из представленной схемы потенциальных поверхностей ясно, ■что разделение систем на жесткие и динамические по существу условно. Процессы, характеризующиеся барьером порядка 20—30 ккал/моль, иногда относят к изомеризациям, а иногда к конформационным превращениям.
Химическая классификация динамических систем
До сих пор мы ограничивались формальным описанием реакций внутримолекулярного обмена, ие привлекая химических соображений. Однако во многих случаях химики используют определенные термины для обозначения внутримолекулярных процессов. Обычно химическая классификация основывается на представлении о механизме процессов и включает интерпретацию переходного состояния. Строго говоря, структура переходного состояния не может быть определена экспериментально, так как это состояние не является стационарным состоянием системы. Тем не менее обширный опыт в этой области позволяет выработать ряд важных эмпирических закономерностей. Рассмотрим некоторые наиболее типичные случаи внутримолекулярной динамики.
Свободное вращение вокруг простых: связей С—С. В подавляющем большинстве случаев барьеры вращення вокруг простой С—С-связи
![]()
(вращение изображено круговой стрелкой) невелики (Кб = 3—5 ккал/моль), так что вращение происходит свободно во всем доступном диапазоне температур.
Заторможенное вращение вокруг С—С-связей при наличии стерических затруднений. В ряде случаев пространственные взаимодействия между валентно несвязанными атомами и группами затрудняют достижение переходного состояния
=-
с /
Пр.и наличии объемистых заместителей R барьер вращения может достигать 10—15 ккал/моль.
Вращение вокруг частично двойных связей. Повышение барьера вращения наблюдается для сопряженных- углеводородов, акролеинов (до 10 ккал/моль):
Наиболее ярко частично двойная связь проявляется в случае амидов карбоновых кислот, сульфоамидов
/
где барьеры вращения достигают 20 ккал/моль.
Инверсия около атома азота. Этот процесс наблюдается в ам*
миаке
и некоторых аминах и амидах (AG*,= 6—10 ккал/моль). В отличие от процессов вращения вокруг связей инверсия осуществляется через плоское переходное состояние.
Инверсии в циклических системах. Типичными примерами систем, испытывающих инверсию цикла, являются циклогексан, пиперидин, диоксан и т. д. Например, инверсия цикла в цикло
не
гексане сопровождается изменением ориентации протона (аксиальный протон переходит в экваториальный и наоборот):
На
Не
»-![]()
![]()
Как правило, инверсии циклов характеризуются барьерами 6—12 ккал/моль. Часто переходное состояние в реакции инверсии цикла можно представлять как результат нескольких последовательных вращений. Полный механизм такой инверсии может включать образование промежуточных метастабильных состояний.
Таутомерия. Обычно процессы таутомерии связывают с такими процессами, в ходе которых происходит изменение валентных состояний отдельных атомов. Например, в процессе кето-енольной таутомерии в р-дикетонах:
Н
I
С С
/1\ / \
С С С с
II % II I
ООО о /
н
изменяется валентное состояние (гибридизация) центрального атома углерода. Таутомерные процессы могут иметь самую различную природу и существенно различаться по барьеру активации. Кето-енольная таутомерия указанного выше типа характеризуется барьерами выше 20 ккал/моль и является медленной в шкале метода ЯМР.
![]() Миграции.
Распространенными примерами являются
миграции водорода (гидридные сдвиги)
в бензолониевых катионах
Миграции.
Распространенными примерами являются
миграции водорода (гидридные сдвиги)
в бензолониевых катионах
= С^к'
(барьеры — 15 ккал/моль); а также в циклопентадиенах
н*
. (j). — 0^н‘
(барьеры ~ 24 ккал/моль).
«Текущие молекулы». Во многих случаях процесс внутримолекулярной перестройки становится чрезвычайно сложным. Потенциальная поверхность тогда представляет собой своеобразный «сильно изрезанный горный рельеф» с большим количеством минимумов и перевалов. Динамическое поведение таких систем иногда представляют себе как непрерывный «поток структуры» (отсюда название «текущие молекулы» от англ. fluxional molecules). Примером «текущей молекулы» является удивительный углеводород — бульвал'ен. Ниже приведен один «акт обмена» в бульва- лене:
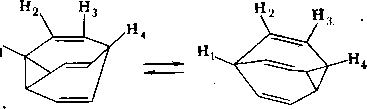
Этот акт включает только две формы. Всего же по расчетам в бульвалене имеется 1 209 600 различных форм, каждой из которых соответствует локальный минимум на потенциальной поверхности.