

§ 2. Химические сдвиги протонов
Определение
Химические сдвиги протонов измеряются в м. д. относительно сигнала тетраметилсилана как внутреннего стандарта (6-шкала ТМС):
6Н
= v»_
-™с..
10*. (3.7)
Vo
Иногда используют т-шкалу (ТМС):
т= 10,00—6. (3.8)
Эмпирические данные
Химические сдвиги протонов занимают диапазон 0—10 м. д. (6-шкала). Стандартная ошибка измерения сдвигов протонов равна ±0,01 м. д.
Химические сдвиги протонов распространенных растворителей и стандартных веществ, дающих один сигнал в спектре, приведены в табл. 3.3.
Таблица
3.3
Химические
сдвиги протонов в некоторых органических
соединениях
Соединения
Формула
6,
м. д.
Циклопропан
С3Н6
0,22
Этан
СНз-СНз
0,88
Циклогексан
СбН12
1,44
Ацетон
СНзСОСНз
2,17
Диметилсульфоксид
(CH3)2SO
2,50
Ацетилен
нс=сн
2,88
и-Диоксан
с4н8о2
3,56
Хлористый
метилен
СН2С12
5,30
Этилен
сн2=сн2
5,84
Хлороформ
СНС13
7,27
Беизол
с8н6
7,27
Трифторуксусиая
кислота
CF3COOH
11,34
Химические сдвиги протонов характеристичны. Для наиболее распространенных функциональных групп химические сдвиги протонов приведены в табл. 3.4.
65
Протоны некоторых функциональных групп (ОН, NH, SH) обнаруживают сильную зависимость от растворителя, концентрации и температуры образца.
Н. М. Сергеев
Таблица
3.4
Химические
сдвиги ‘Н для некоторых структурных
фрагментов
Метильная
группа
в,
м. д.
Метиленовая
группа
6,
м. д.
СНз^С
0,8—1,9
СН2
трехчленный цикл
0,-3—0,9
СНз—С
=
1,6—2,7
СН2
другие циклы
1,2—2,5
СНз—С
=
1,8—2,1
—СН2—
1,2—1,4
СНз—S
2,0—2,6
—СН2—Hal
(кроме
F)
1,3—1,9
СНз—Аг
2,1—2,8
-СН2—СО
2,1—2,4
СНз—N
2,7-3,1
—ch2-n
2,1—2,8
СНз—О
3,2—4,0
—СН2—Аг
2,6—3,0
-сн2—о—
3,4—4,4
СН3
= С— (несопряж.)
4,6—5,0
СН2=С—
(сопряж.)
5,3—5,7
Метановый
протон
6,
м. д.
Прогоны
прн гетероатомах*
6,
м. д.
-сн-
1,4—1,9
R—ОН
1,0—4,5
—сн—со
2,4—2,7
Аг-ОН
4,3—8,0
сн=с—
2,4—3,1
еиолы
.
15—19
—СН—Аг
2,8—3,1
К—SH
1,2—2,1
—СН—S
3,1—й,2
Аг—SH
3,0—3,0
—СН—О
3,8—5,1
NH—
(амины)
10,5—3,3
—СН-На1
4,0—4,2
Аг—NH—
2,9—4,7
—СН=
(несоп-
5,2—5,7
—NH—
(ямиды)
5,4-8,1
ряж.)
+
—СН=
(сопряж.)
5,7—6,7
RNH3
7,7—8,8
—СН=
(аромат.)
6,1—9,3
R—СООН
9,2—13,2
—сн=о
9,3—10,1
—СН—
мостико-
2,1—2,5
вый
Химические
сдвиги сильно зависят' от природы
растворителя, температуры, концентрации.
Закономерности
Для интерпретации химических сдвигов протонов привлекаются различные теоретические концепции. Несмотря на то что эти концепции не дают строгого количественного описания экспериментальных данных, они полезны при проведении качественной интер-' претации.
Разбиение наблюдаемого значения химического сдвйга на отдельные вклады. В основе интерпретации химических сдвигов протонов лежит предположение о том, что химический сдвиг может 6bifb представлен как сумма вкладов, которым соответствуют независимые электронные токи, индуцированные внешним магнитным .полем. Согласно предположению Сайка и Слигстера, экранирование рассматривают как сумму трех вкладов:
сг—СГд.л + СГп.л+Он.л, (3.9)
где величины ад.л, оп.л и о„.л соответствуют трем разным токам. Первый член в (3.9) отвечает диамагнитному току, обусловленному электронами данного атома, поэтому его называют локальным диамагнитным экранированием. Второй член соответствует локальному парамагнитному току. Наконец, третий член, называемый нелокальным экранированием, включает токи, обусловленные другими электронами системы. В принципе, этот член соответствует нндуцированным токам, охватывающим несколько атомов в одной
'и*
|н
"!
I
jr
|
Hj
I
снч
Ih:
1
0
10
1
I
20
I
|
JO
|
i6
1
1
I
I
tmcJ
I
х
Al
1
L
12
10
6
2
0
(У-
шкала
Рис.
3,1. Шкалы протонного экранирования: а
— шкала абсолютного экранирования
для Н+, Н, Н~, Н2
и СН4;
б — 6-шкала ТМС химических сдвигов
'Н
и той же молекуле или в различных молекулах. В последнем случае вклад в экранирование называют межмолекулярным.
Другие типы разбиения общего экранирования на отдельные вклады основываются на выделении специфических взаимодействий, которые можно представить себе в виде независимых эмпирических факторов.
Влияние электронной плотности (о3аряЛ). Обоснованием для введения этого вклада служат два аргумента: 1 — зависимость диамагнитного вклада от плотности электрона на ls-орбитали атома водорода; 2 — корреляции химических сдвигов протонов с электроотрицательностью соседних групп (заместителей).
Как известно, диамагнитное экранирование в атоме водорода составляет 18 м. д. Пренебрегая электрон-электронными взаимодействиями, можно заключить, что экранирование в анионе Н~ будет равно сумме вкладов каждого из электронов, т. е. о(Н_) ^36 м. д. На основе этих оценок можно ввести шкалу абсолютного экранирования (рис. 3.1). В этой шкале химический сдвиг протонов в молекуле Н2, рассчитанный с помощью достаточно точных методов, равен примерно 26 м. д. К сожалению, шкала абсолютного экранирования не имеет практического значения, поскольку ни Нг, ни тем более Н и Н~ неприменимы в качестве стандартов химического сдвига. Переход в общепринятую шкалу химических сдвигов можно провести, если воспользоваться экспериментальным значением сдвига Н2 (или СН4) в б-шкале ТМС.
Пользуясь приведенными выше данными, можно перевести шкалу химических сдвигов в шкалу плотности электронов на ls-орбитали:
p'(ls) ='31 м-д~ :. •■б(м -Ч, (3.10)
18 м. д.
Соотношение (3.10) часто выражают в форме эмпирического правила:
уменьшение электронной плотности около рассматриваемого атома водорода приводит к увеличению химического сдвига соответствующего протона.
Химические сдвиги протонов коррелируют с электроотрицательностью заместителей. Многочисленные данные по этому вопросу часто выражают в форме следующего правила:
для фрагмента СНХ—СН с ростом электроотрицательности заместителя X ближайший протон смещается в слабые поля (а-эф- фект), удаленный протон смещается в более сильные поля (ft-эффект).
При этом предполагается, что электроотрицательные заместители приводят к понижению общей электронной плотности на атоме С 8 и к повышению плотности на атоме Ср.
Вклад магнитно-анизотропных групп. Основанием для введения вклада магнитно-анизотропных групп в экранирование является ряд «аномалий», необъяснимых в рамках корреляций с электронной плотностью, а именно: 1) значение химического сдвига протонов бензола; 2) химические сдвиги протонов ацетилена и циклопропана; 3) химические сдвиги протонор, расположенных вблизи от некоторых связей (С—Hal, С = 0, С—С и т. д.).
В основе интерпретации этих наблюдений лежит модель маг- нитно-анизотропной группы, впервые предложенная Мак-Коннелом.
В частном случае двухатомной молекулы А—В магнитный момент, индуцированный связью АВ при наложении поля Нй, будет различным в зависимости от того, как ориентирована связь АВ относительно поля Но. В простейшем случае аксиальной симметрии, к которому относится молекула А—В, вводят две восприимчивости
(вдоль связи) и x_l (перпендикулярно связи), так что
Ai ,| = х II Но, (3.11а).
М± = Х±Яо- (3.116)
Индуцированный магнитный момент М( = хНо) создает допол-
нительное магнитное поле на соседних (к данной связи) ядрах. Если рассматривать индуцированный момент как диполь и поместить этот диполь в центр связи АВ, то тогда поле можно рассчитать по формуле для поля диполя (1.40).
Поскольку раствор предполагается изотропным, то при расчете дополнительного поля следует провести усреднение по ориентациям связи АВ относительно поля Н0. Свободное вращение молеку-
лы АВ относительно направления поля Я0 равносильно усреднению по трем ориентациям вдоль осей х, у, z, так что среднее поле Н определится из выражения
Н = ± (Н (Мх) ^ Н (Мв) -г Н {Мг)). (3.12)
Используя формулу (1.40), легко убедиться в том, что это поле равно нулю, если %„ =Хх>'Т. е. если группа магнитно-изотропна. При наличии анизотропии Ах=Хи~~Хх =7^0 дополнительное поле оказывается неравным нулю. Учитывая пропорциональность этого поля величине внешнего поля Я0, получим для дополнительного экранирования
Ар
— Ах -Lz3cos*9 (3.13)
3R3
где R — расстояние от диполя до рассматриваемого ядра; 0 — угол, образованный вектором R и осью аксиальной симметрии группы (рис. 3.2). Из формулы (3.13) очевидно, что конус с 0 = 55°44' (рис. 3.2) разделяет области с Ао>0 и ДоСО. Если Ах>0, то протоны, находящиеся внутри конуса экранирования, будут испыты-
Рис.
3.2. Лабораторная система координат
(х',
if,
z')
и
система координат, связанная с
главными осями магнитно-аии- зотропной
группы (х,
у, г).
Поле на ядре Н, индуцированное
магиитно- анизотропной группой,
определяется значениями координат i?
и
0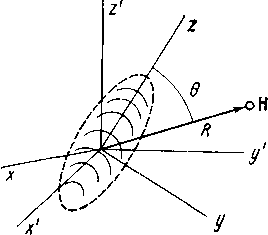
вать сильнопольное смещение, в противном случае сильнопольное смещение будут испытывать протоны, находящиеся вне конуса. Разумеется, чем ближе протон к магнитно-анизотропной группе, тем сильнее смещение.
Частным, но весьма распространенным случаем магнитно-анизо- тропных систем являются ароматические соединения, представляющие собой плоские кольца с сопряженными двойными связями, что приводит к обобществлению 2р-электронов. В таких системах под влиянием магнитного поля возникает прецессия электронного тока в плоскости кольца (кольцевые токи), что и приводит к экранированию соседних протонов.
На рис. 3.3 схематически- изображены конусы экранирования для некоторых химических связей и молекулярных фрагментов.
Рис.
3.3. Конуса экранирования для типичных
магнитио-анизотропных групп и связен.
Области ( + ) и (—) соответствуют областям
экранирования (—б) и дезэкранирования
( + 6)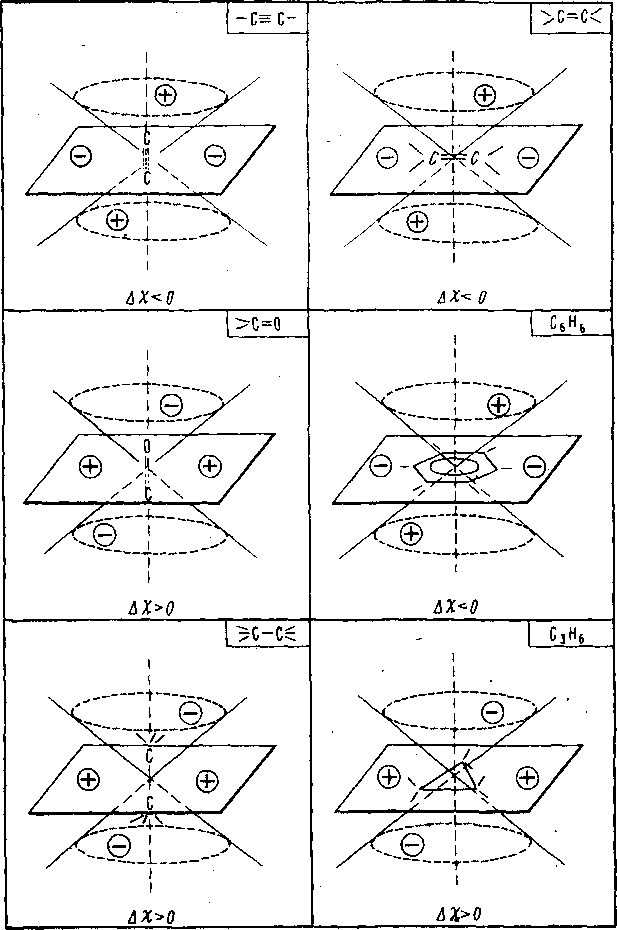
Специфическое влияние растворителя. Под специфическим влиянием растворителя (среды) понимают образование комплексов или ассоциатов, в которых относительное расположение молекул растворенного вещества и растворителя обеспечивается химическими связями. Это могут быть слабые взаимодействия, например, донорно-акцепторное взаимодействие, водородная связь, стекинг (или пачечное) взаимодействие и т. д. Как правило, специфическое влияние растворителя обнаруживается по сильной зависимости химических сдвигов от температуры и концентрации раствора. Наиболее сильно оно проявляется на протонах, участвующих во внутри- и межмолекулярных водородных связях (ОН'... О, NH ... N, SH ... S и т. д.). В этом случае специфический вклад среды может достигать ~10 м. д.
Неспецифическое влияние среды. Сюда относят группу сравнительно слабых эффектов, обусловленных физическими причинами. В частности, если растворитель магнитно-анизотропен, то он может вызывать дополнительные сдвиги на протонах растворенного вещества (иногда эти сдвиги называют ASIS-эффектами от английского anisotropic solvent induced shifts).
Кроме того, к неспецифическим относят дисперсионные (или ван-дер-ваальсовы) взаимодействия. Эти взаимодействия определяют ближний порядок (агрегацию) молекул раствора (т. е. в конечном счете сам факт существования конденсированной фазы). Агрегация молекул приводит к появлению дополнительного экранирования, обусловленного влиянием электронных оболочек соседних молекул. В случае протонного магнитного резонанса роль дисперсионных взаимодействий, по-видимому, невелика, однако для других ядер важность этого вклада убедительно доказана.
Пример вычисления химических сдвигов протонов
Во многих случаях существенный практический интерес представляет следующая задача: известна структура органического соединения, содержащего протоны, требуется рассчитать химические сдвиги этих протонов.
К сожалению, квантовохимические методы решения этой проблемы на практике оказываются малоэффективными. Поэтому в большинстве случаев приходится использовать чисто эмпирические подходы, основанные на привлечении различных модельных соединений, аналогий, корреляционных уравнений, аддитивных схем и т. д.
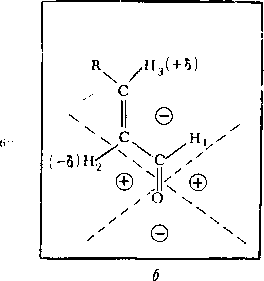
В качестве примера рассмотрим различные эмпирические способы расчета химических сдвигов протонов в транс-гексен-2-але, I
Нч / СН2—СНа—СН3
сон>С=С<н
Для того чтобы считать структуру соединения I заданной полностью, требуются сведения о конформационном состоянии соединения. Для определенности будем считать, что транс-гексен-2-аль находится в s-транс-конформации по
альдегидной группе. Вращение вокруг простых связей С—С считается свободным. Для наглядности рассматриваемая структура приводится иа рис. 3.4.
В результате быстрого вращения вокруг простых связей С—С химические сдвиги геминальных протонов усредняются. Таким образом, в рассматриваемом соединении имеется всего шесть химически неэквивалентных групп протонов Hi—Н6 (рис. 3.4).
 Использование
таблиц и диаграмм химических сдвигов.
Простейшие
оценки химических сдвигов можно провести
с помощью данных, приведенных в табл.
3.4. Однако полученные таким образом
значения (табл. 3.5, способ 1) обнаруживают
значительные разбросы, что не позволяет
рассматривать этот
Использование
таблиц и диаграмм химических сдвигов.
Простейшие
оценки химических сдвигов можно провести
с помощью данных, приведенных в табл.
3.4. Однако полученные таким образом
значения (табл. 3.5, способ 1) обнаруживают
значительные разбросы, что не позволяет
рассматривать этот
Рис.
3.4. Структур» транс-гексен-2-аля;
группа СНО находится в s-транс-
конформации; алкильный радикал свободно
вращается вокруг С—С-связей (а); схема,
поясняющая влияние магнитной анизотропии
карбонильной группы (6)
способ как достаточно удовлетворительный. Рассмотрим, • в частности, химические сдвиги олефиновых протонов Нг и Н3. Оба «расчетных» значения лежат в диапазоне 5,7—6,7 м. д. и, таким образом, не различаются между собой. Попытки качественно учесть влияние заместителей на химические сдвиги этнх протонов также малоэффективны. Так, с одной стороны, можно ожидать, что протон Н2 даст сигнал в более слабом поле, чем Н3, в силу большей электроотрицательности группы СНО по сравнению с CH2R. Однако, с другой сторону, магнитно-анизотропная группа СНО должна приводить к сильнопольиому смещению сигнала Н2 (рис. 3.4,6). Таким образом, речь идет о противоборствующих тенденциях, и для того, чтобы сделать какие-либо выводы, необходимы количественные оценки эффектов магнитной анизотропии и электроотрицательности.
В качестве более солидной эмпирической основы возьмем данные, приведенные в справочнике «Спутник химика» [3]. Этн данные позволяют существенно уточнить химические сдвиги протонов Н,, Н4, Н5 и Н6, одиако оценки для олефиновых протонов и в этом случае оказываются недостаточно точными (табл. 3.5, способ 2).
В настоящее время наиболее обстоятельной компиляцией данных по протонным химическим сдвигам считается руководство Чемберлейна [4], в котором систематизированы данные более чем о 10 000 соединений. Используя данные, приведенные в этом руководстве, можно несколько улучшить точность оценок (табл. 3.5, способ 3). Однако н в этом случае точность химических сдвигов олефиновых,протонов недостаточно велика.
Использование аддитивных схем расчета химических сдвигов протонов. Одна из таких схем, предложенная Примасом и модифицированная Боммером [5], основана па учете ближайшего окружения посредством введения вкладов

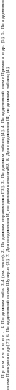
от разветвлений углеродной цепи и от отдельных заместителей. Рассчитанные значения (табл. 3.5, способ 4) не противоречат оценкам, полученным с помощью данных Чемберлейна, за исключением олефинового протона Н3. Этот факт, по-видимому, отражает неудовлетворительность схемы Примаса в отношении расчета химических сдвигов олефиновых протонов. Эти недостатки призвана устранить аддитивная схема Паскуаля и др. [7], разработанная специально для расчета химических сдвигов олефиновых протонов. Точность полученных значений (табл. 3.5, способ 5) близка к ±0,2 м. д., что позволяет рассматривать схему Примаса как непригодную для олефиновых протонов. Наконец, химические сдвиги протонов Н4, Н5, Нб можно оценить с помощью аддитивной схемы Шулери [3], которая используется для расчета сдвигов метиленовых протонов. Полученные с помощью этой схемы оценки (табл. 3.5, способ 6) совпадают в пределах ±0,2 м. д. со значениями, которые получаются из таблиц Чемберлейна.
Использование модельных соединений. Модельное соединение должно удовлетворять двум условиям: во-первых, оно должно быть аналогичным рассматриваемому соединению в отношении всей структуры или какого-то фрагмента структуры; во-вторых, химические сдвиги протонов модельного соединения должны быть известны. Указанным условиям удовлетворяют соединения
Нч /СН3 ,С--=С' НгС=СН-~СН2—СН"2—сн3
СНО/ чн
II III
гранс-Кротоновый альдегид II моделирует химические сдвиги протонов Нь Н2, Н3, Н4, а пентен-2 III — химические сдвиги протонов Н4, Н5, Н6. Соответствующие экспериментальные данные для II и III заимствованы из справочной литературы (табл. 3.5, способы 7 ц 8). Следует оговориться, что, вероятно, наименее точные оценки при использовании указанных моделей получаются длй протона Н4, находящегося на границе двух структур.
Суммируя проведенные выше оценки, можно прнйти к следующим выводам. По-видимому, погрешность эмпирических методов расчета химических сдвигов протонов составляет не менее 0,1 м. д. Для альдегидного протона Hi погрешность возрастает до ±0,5 м. д., вероятно, вследствие температурной и концентрационной зависимости химических сдвигов.