

Photochemistry_of_Organic
.pdfEnergy Transfer, Quenching and Sensitization |
65 |
not generally accepted for more than 30 years. In retrospect, this is a classical example of the Babylonian information barrier between various disciplines of science. Unfortunately, Kautsky had originally attributed the chemically active oxygen species to the then known (upper) excited singlet state of O2. His hypothesis was subsequently buried by the observation that photooxidation could be induced by infrared irradiation beyond 1.31 mm 1, despite the fact that the existence of a lower singlet state had been predicted both by Mulliken and H€uckel and identiÞed by Herzberg in 1934. Kautsky s reassignment of the active species to the lower-lying singlet state in 1939 had little impact. His hypothesis was revived only when Kasha reported on the luminescence of chemically produced singlet oxygen in 1963,132 and when Foote showed a year later that the chemical reactivity of singlet oxygen generated chemically was identical with that formed by sensitization.133
An MO diagram of the dioxygen molecule is shown in Section 4.4, Figure 4.13. The highest occupied orbital is twofold degenerate and these two MOs allocate two electrons in total. Thus, the ground state of molecular oxygen is a triplet state, in accord with Hund s rule. The term symbol for triplet oxygen is 3Sg . Two low-lying singlet states of oxygen,
1Dg and 1Sgþ , have energies of 95 and 158 kJ mol 1, respectively, above the triplet ground state.134Ð136 The upper excited 1Sg þ state is rapidly deactivated in solution to the long-
lived 1Dg state, which is commonly referred to as singlet oxygen, 1O2. Singlet oxygen is an extremely reactive (Section 6.7) and highly cytotoxic species.
Molecular oxygen (dioxygen), 3O2, plays a dominant role in photochemistry. Its concentration is on the order of 2 10 3 M in air-saturated organic solvents at ambient temperature, 0.27 10 3 M in water and 5 10 3 M in perßuoroalkanes, close to that in
air, 8 10 3 M.137 Oxygen diffusion in low-viscosity organic solvents is very rapid, kd 4.5 1010 M 1 s 1 in acetonitrile137 and 2.7 1010 M 1 s 1 in cyclohexane.138 Because
triplet oxygen has two low-lying excited singlet states, it is a very efÞcient quencher of excited states. An excited sensitizer molecule 1S can produce two molecules of singlet oxygen by the sequence of reactions shown in Equation 2.52:
1S* þ 3O2 ! 3S* þ 1O2 or 1S* þ 3O2 ! 3S* þ 3O2
3S* þ 3O2 ! 1S þ 1O2
Equation 2.52 Quenching of singlet and triplet states by dioxygen
Both reactions shown on the Þrst line are spin allowed and may occur on every encounter of 1S with 3O2 (singlet quenching). The Þrst reaction is exothermic only if the singletÐtriplet energy gap DEST of the sensitizer is sufÞcient for energy transfer to 3O2, ES Ð ET 95 kJ mol 1. In that case, formation of 1O2 will be favoured by the energy gap law. If DEST is smaller, singlet oxygen will not be formed, but the second reaction called oxygen-catalysed ISC is always possible. It is spin allowed, because the nascent encounter complex of 3S and 3O2 can have overall triplet multiplicity. The reaction in the second line (triplet quenching) is spin allowed if the encounter complex of 3S and 3O2 has overall singlet multiplicity, as will be the case in every ninth encounter (see Scheme 2.5, Section 2.2.1). Quantum yields of singlet oxygen formation are usually lower than expected from the above simple considerations due mainly to charge transfer interactions in polar solvents.139 In practice, oxygen quenching in aerated organic solvents limits the
66 |
Photophysics and Classification of Primary Photoreactions |
lifetimes of excited organic molecules to about 20 ns for S1 and 200 ns for T1. The quantum yield of any photoreaction that proceeds via an intrinsically longer-lived state will thus be reduced accordingly unless oxygen is removed.
The quantum yields of formation and the lifetimes of singlet oxygen under various conditions have been investigated in considerable detail. The yield of 1O2 is usually determined by chemical trapping with, for example, 1,3-diphenylisobenzofuran (Section 6.7). The lifetime of 1O2 is measured mostly by its phosphorescence emission at 1270 nm,
which has recently been observed in single cells.140 It can also be detected by its absorption to the upper singlet state, 1Dg ! 1Sgþ , n~max ¼ 0.52 mm 1.141 Systematic studies have shown that both the quantum yields142 and the lifetimes143Ð145 of 1O2 can be
understood in terms of the energy gap law (Section 2.1.5) for the dissipation of excess energy to the solvent. The lifetime of 1O2 in D2O (tD 68 ms) is about 20 times longer than that in H2O (tD 3.5 ms) in the absence of quenchers, because the OÐH stretch vibration is a much better accepting mode for the conversion of the electronic excitation energy of 1O2 to vibrational energy of the solvent.
The vibrational deactivation of 1O2 has been elegantly exploited to achieve highly stereoselective photooxygenation of alkenes in which one site is ÔprotectedÕ by a nearby CÐH bond acting as a quencher.146 Replacement of the protective CÐH group by CÐD signiÞcantly reduces the selectivity of attack. The authors anticipate that this new concept of stereocontrol may prove to be general and may facilitate chiral control in a variety of phototransformations.
Special Topic 2.4: Barometric paint
Pressure-sensitive paint (PSP) provides a visualization of two-dimensional pressure distributions on airfoil and model automobile surfaces for wind tunnel research. The airfoil surface is coated with a polymer Þlm that is permeable to oxygen and contains a luminescent pigment. Platinum porphyrins are mostly used, which emit intense phosphorescence that is strongly quenched by oxygen.147 The surface is irradiated in the wind channel and monitored by a CCD camera (Section 3.1). Complementary Þlters are used with the light source and camera to ensure that no light can pass directly from the lamp to the detector. PSP then gives a continuous map of the oxygen pressure on the surface as a function of wind speed using a SternÐVolmer-type relationship for luminescence quenching (Section 3.9.8). A complementary method is based on porphyrin-sensitized singlet oxygen emission at 1270 nm, which can be imaged with an InGaAs near-infrared camera.148
The conventional method used to design and optimize the shapes of airfoil and other surfaces is to construct a Ôloads modelÕ by drilling several hundred small holes called pressure taps, each connected by its own tube to multiplexed pressure transducers. These pressure taps then provide the map of the lift needed to engineer properly the support structure and other features of an airplane. Such models can cost millions of dollars and are time consuming to build. The goal of PSP is to obtain the same information using highly resolved digital images of luminescence with only few pressure taps to calibrate the PSP emission. Furthermore, PSP can provide data from places where it is impossible to install taps, for example, at very thin parts of a wing model.
A Classification of Photochemical Reaction Pathways |
67 |
2.3 A Classification of Photochemical Reaction Pathways
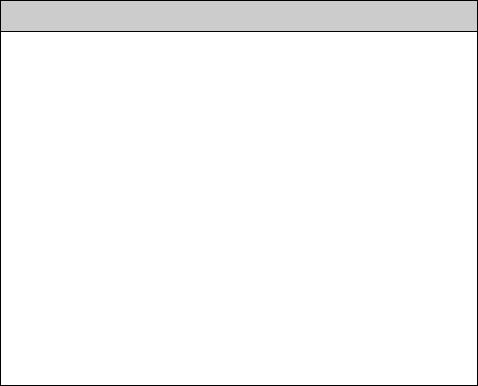
The concept of potential energy surfaces (PES) (Section 1.4) allows us to visualize what should properly be described by the abstract quantum mechanical notion of a wave packet moving in the (3N 6)-dimensional space of the internal nuclear coordinates. The absorption of a photon by a molecule may initially place it on an elevated point of the excited state surface (Franck–Condon principle). Nearby minima on the excited state PES are called spectroscopic minima, because their position and shape can be gleaned from the vibronic structure of the absorption spectrum. The energy released when the molecule starts Ôrolling downÕ on the excited-state surface is irreversibly dissipated to the surrounding medium, much like the potential energy of a rolling marble is eventually converted to heat by friction. Like a marble, the molecule is likely to follow the path of steepest descent.q Having arrived at a local minimum, it is presented with the choice of various photophysical processes, ßuorescence, internal conversion or intersystem crossing. In principle, this choice is always available, but thermal relaxation is fastest on a slope of the PES. Given sufÞcient time near the minimum, the molecule may be jostled over a barrier by thermal reactivation to Þnd a deeper minimum on the excited PES. Once it has arrived on the ground-state surface, the shape of the latter will govern what becomes of it next. The task of theoretical calculations and models (Section 4.9) will be to
Figure 2.26 Potential energy surfaces. Reproduced by permission from ref. 16. Copyright 1990, John Wiley & Sons, Ltd
predict or estimate the shapes of the relevant PES (Figure 2.26) as a guideline to understand the photochemical reactivity of organic molecules.
q This classical analogy should obviously be taken with a pinch of salt. Apart from ignoring wave mechanics, it does not properly account for solvent effects, which may impose a barrier on ÔdownhillÕ changes of molecular structure (see, e.g. Section 5.5).

68 |
Photophysics and Classification of Primary Photoreactions |
|
||||||
|
a |
b |
|
|
|
c |
d |
|
hν |
|
hν |
|
B* |
hν |
|
hν |
|
|
|
–hν |
|
|
|
|
|
|
A |
B |
A |
B |
|
A |
B |
A |
B |
hot ground state |
adiabatic |
|
|
diabatic |
reaction via |
|||
reaction |
reaction |
|
|
|
reaction |
intermediate |
||
Figure 2.27 Four classes of photoreactions
At which point along the reaction coordinate does the molecule jump from the excited state PES to the ground state? Asking this question leads to a classiÞcation of photoreactions to four different categories that were originally proposed by F€orster. Figure 2.27 shows four two-dimensional cross-sections of an excitedand a ground-state PES along a reaction coordinate leading from reactant A to product B. We do not specify at this point whether the excited-state surface represents a singlet or a triplet state.
Class a Internal conversion from the local (ÔspectroscopicÕ) minimum in the excited state forms a ÔhotÕ (vibrationally excited) molecule in the electronic ground state. The excess energy initially acquired by IC or ISC is often more than sufÞcient for some reactions to take place. However, vibrational relaxation will cool the molecule within a few picoseconds in solution and, in practice, hot ground-state reactions leading to a new product B rarely compete with vibrational relaxation back to A. The two-dimensional diagrams shown in Figure 2.27 are somewhat misleading in this respect: Movement along most of the many degrees of freedom other than the reaction coordinate will encounter a high barrier. Reaction towards B therefore requires intramolecular vibrational redistribution to channel a large part of the excess energy into the internal degree of freedom that can heave the molecule towards B. In other words, the density of states is low in the region above a barrier on the ground-state surface, so that the barrier is felt even at excess energies well above its height. Another reason why few hot ground-state reactions have been established to occur may be that it is difÞcult to obtain unambiguous evidence for such a reaction path. In gaseous molecules at low pressure and in rare gas matrices at low temperature, on the other hand, vibrational cooling is slow and hot ground state reactions are quite likely to occur.
Class b Reactions that proceed entirely on an excited-state surface, initially forming the Þnal product in its excited state B , are called adiabatic reactions.r Adiabatic reactions are not unusual in the triplet state, because the competing photophysical decay processes are slow, allowing for the passage across substantial barriers on the triplet surface. Singletstate adiabatic reactions, on the other hand, are usually associated with only minor structural changes with very low barriers such as valence isomerization, geometric isomerization and hydrogen or proton transfer.
r The term ÔadiabaticÕ implies that the reaction path does not Ôcross a boundaryÕ, that is, it does not leave the excited state PES. In thermodynamics, the term ÔadiabaticÕ is used in an entirely different context of system boundaries that are impenetrable to heat and material.

A Classification of Photochemical Reaction Pathways |
69 |
The observation of emission from a product B (or tripletÐtriplet absorption by 3B in ßash spectroscopy) upon excitation of a reactant A would seem to be clear evidence for an adiabatic reaction, class b . However, this is true only when excitation of A is possible at a wavelength where B does not absorb. Otherwise, diabatic reactions can easily masquerade
as adiabatic ones due to re-excitation of the photoproduct B by the light source used for excitation of A.149
Class c Diabatic reactionss proceed directly from the excited state to the ground state of the photoproduct via geometries, at which the two surfaces cross (conical intersections, Special Topic 2.5) or nearly cross (avoided crossings, funnels). In such reactions, no intermediates other than the excited state reached by absorption can usually be detected, because the molecule arrives on or near to a cusp of the ground-state surface and immediately proceeds to a stable product.
Are potential energy surfaces allowed to cross, as shown in diagram c ? This has been under dispute for some time. It was argued that BornÐOppenheimer states belonging to the same irreducible representation, that is, to states with spatial wavefunctions having the same transformation properties under symmetry operations of the molecule (Section 4.4), cannot have exactly the same energy, because an exact Hamiltonian operator would
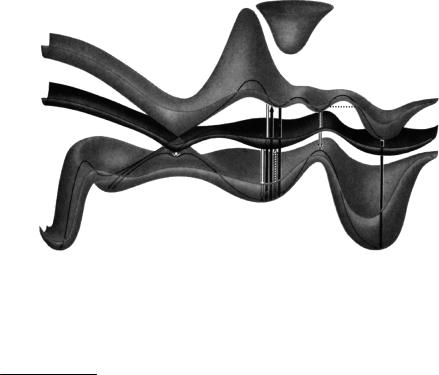
generally give rise to interaction terms that split them apart. In fact, the noncrossing rule holds strictly only for diatomic molecules;150Ð153 surface crossings may occur in a
restricted, (3N 8)-dimensional geometric subspace of polyatomic molecules (see Special Topic 2.5). Nevertheless, consideration of a single reaction coordinate with only one degree of freedom prohibits accidental surface crossings at most geometries, so that only avoided crossings will be seen in most cross-sections of the full (3N 6)-dimensional geometric space such as diagram c . Approximate PESs that cross, because the relevant interaction terms are ignored in a particular model used, are called diabatic surfaces (Section 4.9). Whether a molecule actually descends through a conical intersection or an avoided crossing in a diabatic reaction is, however, a rather academic issue. PESs become meaningless when two states are very close in energy, because the BornÐOppenheimer approximation fails and energy dissipation is very fast in such situations.153
A semiclassical model to calculate the probability p12 that a molecule follows a diabatic pathway, by retaining the electronic structure of the initial electronic state as it passes through an avoided crossing, was developed independently in 1932 by Landau and by Zener.154 In the Landau–Zener model, the nuclear movements are described classically, that is, nuclear motion enters parametrically and only a single reaction coordinate, as shown in diagram c in Figure 2.27, is considered. In the LandauÐZener expression given in Equation 2.53:
p |
|
|
exp |
4p2V122 |
exp |
2 |
|
v |
|
|
¼ |
hyjs1 s2j ¼ |
p |
12tÞ |
|||||
|
12 |
|
|
ð |
|
Equation 2.53 Landau–Zener model for diabatic reactions
s Also called nonadiabatic reactions, a double negation.
70 |
Photophysics and Classification of Primary Photoreactions |
Special Topic 2.5: Conical intersections
Two conditions must be met at a crossing of two PESs: the energy of the two states must be the same and the interaction between the two electronic states must vanish. The electronic interaction between states of different multiplicity is very small in general, so PESs of different multiplicity can cross freely, but the additional requirement of vanishing interaction between states of equal multiplicity reduces the (3N 6)- dimensional space of the nuclear coordinates by a second degree of freedom. Hence PESs are allowed to cross in a (3N 8)-dimensional subspace, even if they have the same spatial symmetry. In linear molecules, there are 3N 5 degrees of freedom and the subspace reduces to 3N 7. Diatomic molecules have only one degree of freedom, the internuclear distance r. Hence both conditions cannot be met simultaneously and states of the same symmetry cannot cross. This is the noncrossing rule for diatomics.
If the PESs are plotted against two special internal coordinates x1 and x2, which deÞne the so-called branching plane, they would have the shape of a double cone in the region surrounding the degeneracy. In the remaining 3N 8 directions the PESs remain degenerate, whereas movement in the branching plane lifts the degeneracy (Figure 2.28).
Figure 2.28 Conical intersection. Adapted from ref. 22
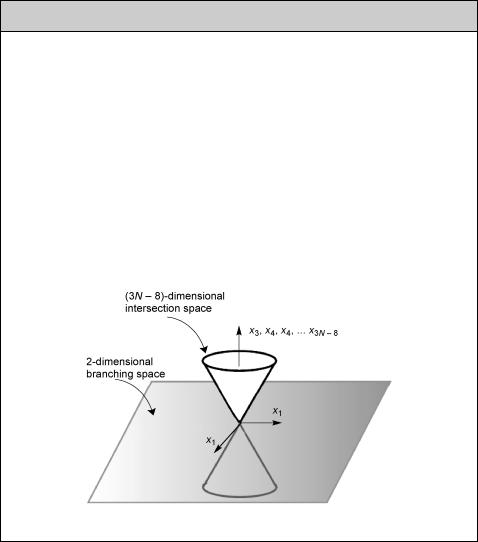
V12 is the electronic coupling term between the zero-order states at the crossing point, h is Planck s constant, y (not n!) is the nuclear velocity along the reaction coordinate as the molecule approaches the crossing point and |s1 Ð s2| is the difference between the slopes of the two intersecting PESs at this point. Even in systems for which an accurate calculation of p12 is possible nowadays, the LandauÐZener model provides a useful Þrst estimate.
Equation 2.53 makes sense when one realizes that 2pV12/h is the frequency v12 with which the system oscillates between the two electronic structures represented by the diabatic states and V12/|s1 Ð s2| represents an Ôinteraction lengthÕ so that the duration t of the interaction equals V12/[y|s1 Ð s2|]. Consider the two cases shown in Figure 2.29.
The zero-order diabatic PESs that cross at the origin of the reaction coordinate are shown as dotted lines and the interaction V12, chosen at 0.05 eV, leads to an avoided
Problems |
71 |
Figure 2.29 Diabatic and adiabatic reaction paths
crossing of the adiabatic curves (solid lines). Atoms move at speeds on the order of 104Ð105 cm s 1, depending on their weight. We choose 105 cm s 1 for relatively light atoms. The slopes of the diabatic PESs are chosen as 0 and 0.01 eV pm 1 on the left and as 0 and 0.05 eV pm 1 on the right. For a molecule moving from left to right along the reaction coordinate, the LandauÐZener model (Equation 2.53) then predicts p12 ¼ 9%, the probability for the diabatic reaction to occur as it passes through the region of the avoided crossing, that is, a 91% probability for staying on the upper PES. When the difference in slope is Þve times higher (right), p12 amounts to 62%.
Diabatic reactions are rarely initiated from triplet states, because the interaction terms V12 between states of different multiplicity, which are due to spinÐorbit coupling (Section 4.8) are small. Even if surface crossings with the singlet ground-state PES do occur in certain regions of the (3N 6)-dimensional space of the internal nuclear coordinates and the molecule does explore such a region in its lifetime, it is unlikely to Þnd the minute escape hatch to the ground state PES.
Class d The reaction proceeds by formation of reactive intermediates such as a radical pair, a carbene, a biradical or a zwitterion, which have low-lying excited states and are therefore situated near a minimum on the excited state PES. Many photoreactions belong to this class (see Section 5.4).
2.4 Problems
1.Using Equation 2.11 and the absorption spectrum of anthracene shown in Figure 2.11,
calculate the ßuorescence rate constant kf for anthracene in a solvent of refractive index n ¼ 1.5. For a rough estimate of the area of the Þrst absorption band, it is sufÞcient to
sketch a rectangle of about the same area and replace the integral by the product of height and width of the rectangle [kf 1 108 s 1].
2.Estimate the rate constants kIC and kISC from S1 and kISC from T1 for anthracene. Refer
to Equation 2.22, Equation 2.23 and Table 8.6. [kI C 5 106 s 1 , kISC(S1) 6 107 s 1, kISC(T1) 1 104 s 1].
3.Referring to the spectra of anthracene (Figure 2.11) and biacetyl (Figure 6.5), give a quick-and-dirty estimate of the critical F€orster distance R0 (Equation 2.37) for FRET
72 |
Photophysics and Classification of Primary Photoreactions |
from anthracene to biacetyl. Is F€orster theory adequate to predict R0 in this case? [R0 50 pm. No, the calculated distance is too short to warrant using the dipole approximation].
4.IR-absorbing dyes with high quantum yields of ßuorescence are hard to come by. Explain. [Low rate constants of ßuorescence (Equation 2.11), fast competing IC (Equation 2.22)].
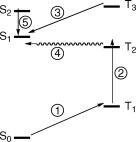
5.Give an adequate name to the processes shown in the Jabłonski diagram (Figure 2.30) and discuss the probability of these processes or any special conditions that may be required to permit their observation. [\: SingletÐtriplet absorption; very weak, heavyatom solvent or oxygen pressure, phosphorescence excitation. `: TripletÐtriplet absorption; ßash photolysis. ´: TripletÐsinglet phosphorescence; unlikely, because IC is fast. ˆ: Triplet-Singlet ISC: unlikely, because IC is fast. ˜: Fluorescence; very weak, violation of Kasha s rule].
Figure 2.30 Photophysical processes
6.In a solution containing 1 10 5 M of a dye, «(lirr) ¼ 2.5 104 M 1 cm 1, what percentage of radiation at lirr is absorbed by the dye, if the absorbance of the solvent in
the absence of dye in a 1 cm cell is A(lirr) ¼ 0.4? [38.5%].
7. Calculate the probability p12 for diabatic reaction (Equation 2.53) given that y ¼ 105 cm s 1, V12 ¼ 0.02 eV and |s1 s2| ¼ 0.02 eV pm 1. [p12 ¼ 83%]
8.On the basis of Arrhenius empirical law for Þrst-order rate constants, kr ¼ Aexp(ÐEa/ RT), calculate the maximum activation energies Ea that permit reactions initiated on a singlet and on a triplet state PES to compete with the rate constants, kd, of the unavoidable photophysical decay processes occurring from these states. Assume
kr(S1) |
¼ |
kd(S1) |
|
1 |
|
108 s 1, |
|
kr(T1) |
¼ |
kd(T1) |
|
1 |
|
103 s 1, |
A |
¼ |
1 |
|
1013 s 1 |
and |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
T ¼ 300 K. [S1: Ea ¼ 30 kJ mol |
1 |
. T1: Ea ¼ 60 kJ mol |
1 |
]. |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|||||||||||||||
9.The quantum yield of some photoreaction increases 100-fold when the solution is
degassed. Which excited state is likely being quenched by dioxygen and what is its lifetime in degassed solution? [T1, 20 ms].

3
Techniques and Methods
Recommended textbooks.137,155–159
3.1 Light Sources, Filters and Detectors
The spectrum of the main light source on Earth, the Sun, was shown in the Introduction (Figure 1.1). It contains only a small amount of UV radiation. Photolyses in the laboratory are carried out using artificial sources of ultraviolet or visible light, combined with optical filters, which confine multiwavelength radiation to the desired spectral region. Compilations of the spectral distributions of various light sources, of cut-on, cut-off,
narrow-band or solution filters and other optical components are available in hand- books1,137,156–158 and from many commercial providers (Oriel). Here, we cover only a
selection of the material.
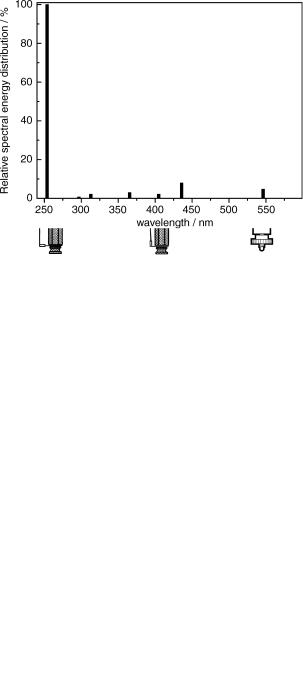
The most widely used sources of UV–VIS light for continuous irradiation in laboratory experiments are xenon and mercury arc lamps, whereas deuterium and tungsten lamps are utilized for spectrophotometers. Various mercury lamps (Figure 3.1) are commercially available. Their spectral irradiance is strongly dependent on the mercury vapour pressure. Figure 3.2 shows the line spectrum of a typical low-pressure mercury arc (the vapour pressure of mercury is about 10 3 mbar). That of a medium-pressure ( 1 bar) lamp is shown in Figure 3.3. The former emits primarily two bands of radiation centred at 253.7 and 184.9 nm, due to Hgð3P1Þ and Hgð1P1Þ de-excitation, respectively. Depending on the transmittance of the quartz envelope, the short-wavelength line may be partially filtered out. These lamps are often utilized in Rayonet reactors (see below). The excited mercury atoms in medium-pressure arc lamps, undergoing more frequent collisions with electrons, are in part excited to higher states. As a result, more emission lines, such as 313.9 or 365.4 nm, useful for photochemical experiments, become available. The lamp envelope s temperature can reach 600 C and produce a considerable amount of infrared radiation and heat. Therefore, cooling-water circulation or water cuvette filters must be utilized to prevent the sample from becoming heated.
Photochemistry of Organic Compounds: From Concepts to Practice Petr Klán and Jakob Wirz © 2009 P. Klán and J. Wirz. ISBN: 978-1-405-19088-6
74 |
Techniques and Methods |
Figure 3.1 Examples of low- (left), medium- (middle) and high-pressure (right) mercury arc lamps. Reproduced by permission of Ace Glass Inc. and Newport Corp, Oriel Product Line
The most intense sources of UV radiation are the high-pressure ( 100 bar) mercury arcs. The spectral lines are broadened due to the high pressure and temperature and they are superimposed on a continuous background of radiation (Figure 3.4). While common mercury–xenon [Hg(Xe)] lamps still produce significant mercury emission bands, especially in the UV region, the smoother xenon lamp spectrum finds application in environmental photochemistry experiments because of its resemblance to solar radiation (Figure 1.1).
Figure 3.2 Relative spectral energy distribution of a 15 W low-pressure Helios Italquartz Hg lamp. Adapted from ref. 157