

Photochemistry_of_Organic
.pdf196 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||||||||||||||||
|
|
|
|
|
∆Go{pH = 0}/(2.3RT) |
|
|
|
|
|
|
|
|
|
|||||
|
55 |
|
|
|
T1 |
|
|
|
|
pKa* = 3.6 |
|
|
|
|
|
pKa* = 4.6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
50 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
pKE(T1) ≈ – 1.0 |
|
|
E0–0 = 47.5 |
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
15 |
|
|
|
|
|
|
E0–0 = 51.8 |
|
|
|
|
pKa = – 8.5 |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
|
10 |
|
|
|
|
|
|
pKa = 7.9 |
|
|
|
|
|
pKE = 16.4 |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
5 |
|
|
|
S0 |
|
|
|
|
|
|
|
calculated |
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
||||||||||
|
0 |
|
|
|
|
|
|
|
O |
|
|
|
|
O |
|
|
|
OH |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
HO |
11 |
O |
|
|
|
11– |
O |
12 |
||||||
Figure 5.8 Protonation equilibria of 4-hydroxyacetophenone (11) in the triplet state
Thus, in the pH range 4–6, the triplet enol tautomer 12 is thermodynamically more stable than the triplet ketone 11. The spectroscopic and thermodynamic data are summarized in Figure 5.8. When the enol 12 decays to the ground state, it becomes an extremely strong acid and it immediately protonates water to give the anion 11 . The pKa of enol 12 in the ground state was estimated at 8.5 on the basis of calculations for the equilibrium between 11 and 12 and the measured triplet pKa (12) ¼ 4.6.
Special Topic 5.2: Isotope effects and tunnelling
Isotopes play a multifaceted role in mechanistic chemistry. Isotopic labelling at specific sites of a reagent is often used to support or exclude a hypothetical reaction mechanism based on the location of the isotopic label in the products. Kinetic isotope effects, that is, changes in rate constants that are induced by isotopic substitution, may be seen in time-resolved experiments or may express themselves as changes in product quantum yields. They hold important information regarding the nature of the rate-determining step that limits the overall rate in a sequence of reactions.
Deuterium isotope effects are studied most often, because deuterium is relatively cheap, the synthesis of deuterium-labelled compounds is frequently straightforward and the effects are large and thus easy to measure with sufficient accuracy. Kinetic isotope effects can be attributed largely to the difference in the zero-point energies of the C H and C D stretching vibrations in the reactant, E0 ¼ hn/2. The vibrational

Proton Transfer |
197 |
frequency n is related to the force constant k of the (harmonic) potential and the reduced mass m of the oscillator (Equation 5.11).
n |
|
1 s; m |
|
X |
m |
Y |
|||
|
¼ |
|
|
k |
¼ |
m |
|
||
|
p |
|
m |
|
mX þ mY |
||||
Equation 5.11 Vibrational frequency of a harmonic oscillator
In Section 2.1.5 (Figure 2.7), we encountered kinetic isotope effects on the rates of radiationless decay, which were attributed to the large difference between the frequencies of C H and C D stretching vibrations. The hydrogen isotopes X being much lighter than the nuclei Y to which they are attached, one can, to a first
approximation, neglect mX ¼ 1u or 2u (X ¼ H or D) relative to mY 12u in the |
|||||
denominator of Equation15.11 to obtain m mX and nH/nD |
p |
|
|||
2. For a C H stretch |
|||||
vibration, ~v |
|
|
1 |
|
1 |
|
3000 cm , the difference in the zero-point energies |
is substantial, |
|||
E0(C H) – E0(C D) hc~vH(1 – 1/p |
2)/2 440 hc cm or |
5 kJ mol . On the |
|||
other hand, the H or D atom will be |
very loosely bound in the transition state of a |
||||
hydrogen-transfer reaction such as photoenolization (Section 6.3.6). The activation energy Ea for D transfer will, therefore, exceed that of H transfer by about 5 kJ mol 1. One can then estimate from the Arrhenius equation, k ¼ Aexp( Ea/RT), that the ratio of the corresponding rate constants, kH/kD, may approach 10 at room temperature and higher values below. Isotope effects kH/kD 5–10 are called primary isotope effects and they indicate that the proton is in flight and therefore loosely bound in the transition state of the observed reaction step. If the role of the isotopically labelled atom is only that of a spectator located near the reaction centre in the transition state, kinetic isotope effects are much smaller, kH/kD 1.0 0.1 (secondary isotope effects).
According to the empirical Arrhenius equation, the logarithm of the rate constant k should fall off linearly with a slope of Ea/RT when plotted against 1/T. However, when the temperature is strongly reduced, one sometimes finds that log(k/s 1) levels
off, as shown in Figure 5.9, where the following Arrhenius parameters were assumed: A ¼ 1 1013 s 1, Ea(X ¼ H) ¼ 6 kJ mol 1 and Ea(X ¼ D) ¼ 10 kJ mol 1. In addition, a
constant term kt was added to the Arrhenius equation, k ¼ Aexp( Ea/RT) þ kt, namely kt(X ¼ H) ¼ 1 1012 s 1 and kt(X ¼ D) ¼ 1 1010 s 1.
Figure 5.9 Arrhenius plot for a reaction with tunnelling

198 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
The temperature-independent term kt is attributed to tunnelling of the hydrogen or deuterium atom through the barrier, a quantum mechanical phenomenon. The rate of tunnelling depends on the shape (the height and width) of the barrier and on the mass of the particle.384 Thus electrons tunnel much more easily than nuclei and H tunnels more easily than D and heavier nuclei. Although tunnelling from a given vibrational level requires no activation energy and its rate is therefore independent of temperature, it does proceed more easily from higher levels, where the barrier width and height are reduced (Figure 5.9). The population of upper levers increases at higher temperature. Hence tunnelling contributions to hydrogen atom transfer rate constants may well be substantial even at ambient temperature.385
A case in point is the photoenolization reaction of o-alkylphenyl ketones (Section
6.3.6) and related compounds. Detailed studies of the temperature dependence of both the primary photoreaction386–388 and of the back transfer reaction in the ground state389
have provided convincing evidence for tunnelling contributions in these reactions.
5.4 Primary Photochemical Intermediates: Examples and Concepts
Recommended textbooks.390,391
The wealth of information on reactive intermediates is covered in textbooks of physical organic chemistry and the specialized books quoted above. Many reactions involving such intermediates will be discussed in Chapter 6.
5.4.1Carbenes
Many photoreactions proceed via carbene intermediates (see Schemes 6.169–6.172). Spectroscopic and theoretical attempts to determine the equilibrium bond angle of the parent carbene, methylene (CH2), in its triplet ground state and the energy gap to its lowest singlet state had long given discrepant results that were resolved only in 1984.392 The triplet ground state of methylene is bent (134 ), but the inversion barrier is low and the singlet–triplet energy gap DEST amounts to 38 kJ mol 1.393 Two valence electrons occupy the two symmetry-adapted NBMOs of CH2, which are a pure p-p and an essentially sp2- hybridized s AO (Figure 5.10). The energy of the p-p AO is substantially higher than that of the sp2-s AO, which has substantial s-character. The equilibrium bond angle is reduced to 102 in the lowest singlet state. The two unpaired electrons are localized on the same
|
sp2-σ |
|
|
|
|
SOC |
ISC |
|
|
p-π |
C |
C |
||
fast |
||||
|
||||
|
|
H |
||
H |
H |
H |
||
|
|
|||
lowest singlet state |
triplet ground state |
|||
Figure 5.10 Ground-state triplet (left) and lowest singlet state (right) configurations of CH2
|
Primary Photochemical Intermediates: Examples and Concepts |
199 |
||
Table 5.1 Geometries and singlet–triplet gaps of substituted carbenes X–C–Y394 |
|
|||
X |
Y |
DEST/kJ mol 1 |
f(X–C–Y) |
|
|
|
|
Singlet |
Triplet |
|
|
|
|
|
F |
F |
236 |
104 |
118 |
Cl |
Cl |
84 |
109 |
126 |
Br |
Br |
52 |
112 |
129 |
H |
F |
66 |
102 |
121 |
H |
Cl |
27 |
101 |
124 |
SiH3 |
SiH3 |
82 |
180 |
180 |
atom, so that the exchange integral favouring the open-shell triplet state is large. Nevertheless, the energy gap DEST of CH2 is rather small, because the closed-shell singlet state is stabilized due to the lower energy of the doubly occupied sp2-s AO. Thus, Hund s rule favouring the triplet state applies.
Whereas the absolute prediction of singlet–triplet energy gaps in carbenes has long been a challenge for theoretical chemistry, the rationalization of trends in substituted carbenes XCY is straightforward.394,395 Two kinds of interactions should be considered; inductive effects affect the energy of the s-AO and conjugative interactions affect the p-p AO. The substituents X and Y may be classified as p-donors ( NR2, OR, SR, halogens) and p- acceptors ( CO R, NO2), which raise and lower, respectively, the energy of the p-p AO. s-Donors ( SiR3) and s-acceptors (halogens, especially F) raise and lower, respectively, the energy of the sp2-s AO. Factors influencing the bond angle such as sterically demanding substituents and geometric constraints in cyclic carbenes should also be considered; bond angles close to 180 favour the triplet state, smaller bond angles near 120 increase the s-character of the s AO and favour the singlet. Based on these criteria, the trends shown in Table 5.1 are easily rationalized.
Intersystem crossing in carbenes is extremely fast, because it is associated with transfer of an electron from the sp2-s AO to the p-p AO, which is the ideal situation for strong firstorder SOC (Section 4.8). It is therefore valid to assume that singlet–triplet equilibration generally precedes intermolecular reactions of carbenes (see Case Study 6.29). Singlet carbenes are frequently more reactive and tend to dominate the chemistry, even when the triplet state is slightly lower in energy. Most bimolecular reaction rates of singlet carbenes are close to diffusion controlled and inert solvents allowing their intermolecular reactions to be studied are not readily available; Freon 113 (1,1,2-trichloro-1,2,2-trifluoroethane) and acetonitrile are often used, but the latter may form ylides with reactive singlet carbenes.396 Singlet carbenes are readily protonated to give carbocations (Section 5.4.5).
Fast intramolecular reactions of singlet carbenes include 1,2-shifts (predominantly H- shifts) forming alkenes and the Wolff rearrangement forming ketenes (Scheme 6.171). In adamantylidene, the 1,2-H shift is impeded by geometric constraints; it therefore represents a convenient system to study intermolecular reactivity of a simple dialkylcarbene;397 it has a singlet ground state.398 Carbenes undergo C H and O H insertion (Scheme 6.178) and they add to alkenes, forming cyclopropanes, a synthetically very useful reaction (see Schemes 6.169–6.173). According to the Skell hypothesis, also known as the Skell–Woodworth rule, singlet carbene additions to alkenes are concerted and stereospecific [addition to a (Z)-alkene gives the cis-substituted cyclopropane

200 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
|||
|
Br |
CF3 |
R |
R |
|
N |
N |
||
|
|
Br |
||
|
|
|
|
|
|
Ph |
Ph |
N |
N |
|
|
CF3 |
R |
R |
|
|
|
||
Scheme 5.5 Persistent carbenes
exclusively], whereas those of a triplet carbene are stepwise, proceeding via a triplet biradical intermediate with sufficient lifetime to permit conformational rotation, which results in scrambling of the stereochemical relationships. Based on this criterion, singlet–triplet energy gaps have been estimated from the product distribution.
Some remarkably persistent carbenes have been synthesized. This was achieved either
by substitution with sterically demanding groups that hinder dimerization (triplet carbenes)399,400 or by substitution with strong p-electron donors (singlet carbenes)401
(Scheme 5.5). The latter are probably better described as ylides (zwitterions).
A striking, but well-understood, exception from the tenet of fast singlet–triplet
equilibration in carbenes is represented by 2-naphthyl(carbomethoxy)carbene (13), Scheme 5.6.212,402–405 Irradiation of the diazo precursor in an argon matrix at 12 K gave a
mixture of two conformational isomers of the triplet carbene 313, which were characterized by IR (2120, 1846, 1660 cm 1), UV–VIS (sharp peak at 590 nm with a 1380 cm 1 progression) and ESR spectroscopy.402 Upon subsequent irradiation at >515 nm, 313 was converted to the singlet carbene 113 that persisted for hours in the dark at 12 K! On standing overnight at 12 K or irradiation at 450 nm, 113 was reconverted to 313. The remarkable persistence of 113 at 12 K is attributed to an associated change in the conformation of the carbomethoxy group. These findings represent a rare, but convincing, example for conformational control of the spin state. The minima on the singlet and triplet PESs lie at different geometries, so that ISC from 113 to the triplet ground state is inhibited by an energy barrier. Pump–probe spectroscopy of the diazo
precursor in solution has shown that nitrogen loss occurs on a subpicosecond time scale and that the rate constant of ISC 113 ! 313 is at least 1011 s 1 at ambient temperature.404
Time-resolved IR studies have further probed the equilibrium between the spin states of
O OMe |
O OMe |
|
|
N2 |
|
|
|
hν (450 nm) |
hν > 515 nm |
O |
|
|
OMe |
||
|
∆ (12 K) or |
||
N2 |
|
||
hν (450 nm) |
113 |
||
OMe |
OMe |
||
|
|||
O |
O |
|
|
|
3 13 |
|
Scheme 5.6 Matrix photolysis of a-diazo(2-naphthyl)acetate in Ar at 12 K402
Primary Photochemical Intermediates: Examples and Concepts |
201 |
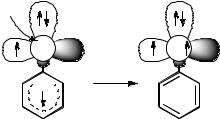
the carbene.212,403 The triplet carbene 313 is the ground state in hexane, but the singlet 113 is the ground state in acetonitrile; it is stabilized by about 4 kJ mol 1 relative to 313 in the more polar solvent. The equilibrated carbenes undergo Wolff rearrangement and C–H bond insertion into the methyl group of the ester.
5.4.2Nitrenes
Recommended reviews.406,407
Nitrenes (R N) are hypovalent species that are commonly formed by thermal or photochemical denitrogenation of azides (R N3), which are readily synthesized and isolated, especially acyl and aryl azides. Simple alkyl azides do not in general produce alkyl nitrenes as detectable or trappable reactive intermediates upon direct irradiation (254 nm) due to rapid 1,2-migration to form the corresponding imines. Trifluoromethylnitrene is, however, formed by irradiation of CF3N3.408
A lucid discussion of the electronic structure of nitrenes has been given407 based on high-quality ab initio calculations that are consistent with experimental data. The ground state of the parent diatomic nitrene, imidogen (NH), is a triplet state. The lowest singlet state is twofold degenerate and lies 150 kJ mol 1 above the triplet ground state. The energy gap DEST is much larger than in methylene (Figure 5.10) because none of the symmetryequivalent 2p-AOs of NH has any s-character. Due to the reduced symmetry of phenylnitrene, its lowest singlet state is no longer degenerate and the gap between the triplet ground state and the lowest singlet state is reduced to 75 kJ mol 1.
The symmetry properties of the spatial wavefunctions describing the lower singlet and the triplet ground state of phenylnitrene are the same (Figure 5.11). The wavefunctions and the relaxed geometries of the two lowest electronic states obtained by ab initio calculations are, however, substantially different. In the singlet wavefunction, the highest, singly occupied p-orbital is almost completely localized in the phenyl ring to keep its electron apart from the other unpaired electron in the p-s AO. This state may be viewed as an iminyl radical attached to a cyclohexadienyl radical. In the triplet state, the same p- orbital is largely localized on the more electronegative N atom because the large exchange integral between the two unpaired electrons keeps the repulsion between electrons of the same spin low. As the lowest singlet and triplet states of phenylnitrene are described by the same orbital occupancy, first-order SOC is zero (Section 4.8). Moreover, the energy gap is
p-π |
sp-σ |
|
p-σ |
N |
N |
|
|
ISC |
|
|
slow |
lowest singlet state |
triplet ground state |
|
Figure 5.11 Ground-state triplet (left) and lowest singlet state (right) configurations of phenylnitrene
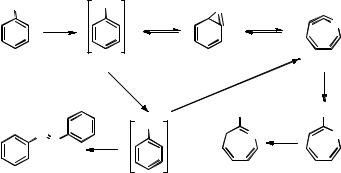
202 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||
|
N3 |
1 |
N |
N |
|
|
hν |
N |
|||
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
14 |
15 |
|
|
|
ISC |
hν |
HNEt2 |
|
|
|
3 N |
NEt2 |
NEt2 |
|
|
N |
N |
NH |
|
|
|
|
|||
|
|
N |
|
|
|
|
|
|
|
16 |
|
|
|
Scheme 5.7 Photochemistry of phenyl azide |
|
||
larger, therefore singlet–triplet ISC in nitrenes is orders of magnitude slower than in carbenes.
The parent phenylnitrene has been studied in detail.406 The thermal or photochemical decomposition of phenyl azide and most of its derivatives in solution results in the formation of intractable polymeric tars. Meaningful mechanistic studies became possible only when it was found that amines intercept the intermediate formed by thermal409 or photochemical410 decomposition of phenyl azide in solution that was responsible for polymerization. The current knowledge about the mechanism of phenyl azide photochemistry is summarized in Scheme 5.7.
The reactions of the primary photoproduct singlet phenylnitrene are strongly temperature dependent.411 At room temperature, ISC is too slow to compete with cyclization to the benzazirine 14 followed by ring opening to the 1-aza-1,2,4,6- cycloheptatetraene (15). The didehydroazepine 15 is trapped by diethylamine, yielding the isolable azepine 16. ISC of the singlet nitrene to the triplet nitrene, which is largely independent of temperature, kISC 3 106 s 1, becomes important only at temperatures below 200 K. All intermediates shown in Scheme 5.7 have been firmly characterized by various spectroscopic techniques, except for the benzazirine 14.406 According to calculations, the ring expansion of singlet phenylnitrene does proceed in a two-step mechanism via 14, but cyclization to 14 is the rate-determining step preceding rapid ring opening to 15. In glassy solutions at 77 K, ISC is the dominant process. At this temperature, the triplet nitrene is stable in the dark, but it is very light sensitive, yielding 15 upon irradiation. Triplet phenylnitrene dimerizes to azobenzene when the glass is thawed. The effects of various substituents on the phenyl ring and the reactivity of naphthyl and other aryl azides have also been investigated.406
Singlet arylnitrenes are strong bases that form nitrenium ions in aqueous solutions.412 The acidity constant of the conjugate acid of 4-biphenylylnitrene has been estimated as pKa ¼ 16. In-plane protonation of singlet phenyl nitrene is, however, a state-symmetry forbidden reaction (cf. Figure 4.32, Section 4.9), because phenylnitrenium ion has a closed-shell singlet ground state.413 Indeed, the protonation of singlet nitrenes by water is substantially slower than would be expected for such a strong nitrogen base and it rarely competes efficiently with ring expansion. Even with strong mineral acid, the rate of

Primary Photochemical Intermediates: Examples and Concepts |
203 |
||||
N3 |
1 N |
1 NH |
NH |
NH2 |
|
|
hν |
H2O |
HO |
|
|
|
H2O |
–HO |
|
|
|
H OH |
|
|
|
OH |
|||
|
|||
Scheme 5.8 |
|
|
|
protonation of singlet 2,4,6-tribromophenylnitrene is about an order of magnitude below the diffusion-controlled limit despite the huge exothermicity of this reaction.414 On the other hand, phenylnitrenium ions are extremely electrophilic and, once formed, they rapidly add water to the para-position in aqueous solution (Scheme 5.8). The lifetime of parent phenylnitrenium ion has been estimated to be 125–240 ps in aqueous solution using an azide trapping method.415
Due to the above-mentioned complications, parent phenylnitrenium has only recently been detected as a transient intermediate using femtosecond pump–probe spectroscopy of phenyl azide in the non-nucleophilic solvent 100% formic acid.416 Phenylnitrenium ion (lmax ¼ 500 nm) is formed with a rise time of 12 ps and has a lifetime of 110 ps in this medium. para-Substituted nitrenium ions have much longer lifetimes. Thus 4- biphenylylnitrenium and 2-fluorenylnitrenium are readily observed by nanosecond flash photolysis of the corresponding azides in water or aqueous acid with lifetimes of about 300 ns and 30 ms, respectively.412 Both of these nitrenium ions react with guanine
derivatives such as 20-deoxyguanosine with rate constants close to the diffusion limit.412,417 These observations may well explain why arylamines that are protected at
the para-position (e.g. 2-naphthylamine) are carcinogenic. Arylamines are known to undergo a twofold metabolic activation yielding nitrenium ions that will attack DNA given that they are sufficiently persistent in aqueous solution.418
Alkylnitrenium ions have also been studied by transient absorption spectroscopy. The antiaromatic carbazolylnitrenium ion is an open-shell singlet that exhibits strong absorption at 620 nm and has a lifetime of 330 ns in acetonitrile solution
(Scheme 5.9).419 Its reaction rate constant with 1,3,5-trimethoxybenzene (TMB) is 9.5 109 M 1 s 1.
Organic azides are used for photoaffinity labelling (see Special Topic 6.16) because the azide group is small and, when attached to enzyme inhibitors, they often retain their activity as inhibitors. Azides are readily synthesized and the light-induced denitrogenation
|
|
|
|
|
OMe |
N |
hν |
N |
TMB |
MeO |
OMe |
N |
BF4 |
|
|
|
N |
|
|
–H |
|
||
|
|
|
|
|
Scheme 5.9 Carbazolylnitrenium ion

204 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
is efficient. Unfortunately, the rapid ring expansion of singlet arylnitrenes is usually too fast to permit the desired cross-linking reactions and triplet nitrenes are not sufficiently reactive to be useful. Various strategies have been followed to increase the lifetime of singlet nitrenes by inhibiting ring expansion. A promising candidate is 2,3,5,6-tetrafluoro- 4-azidoacetophenone, which releases a singlet nitrene with a lifetime of 172 ns in benzene solution that reacts with many substrates to form robust cross-linking.420 Nitrene derivatives that have a singlet ground state, such as acylnitrene,421 and nitrenes forming didehydroazepines (15) that are sufficiently reactive to trap the amine functions of peptides and nucleosides have also been proposed.422 2-Fluorenyl azide, which forms nitrenium ions in aqueous solution, could also be considered for photoaffinity labelling.
5.4.3Radicals and Radical Ions
Numerous photoreactions producing radical intermediates and the products resulting therefrom are discussed in Chapter 6. These reactions include homolytic cleavage such as the a-cleavage of ketones (Norrish type I reaction, Section 6.3.3), hydrogen abstraction by ketones (Scheme 6.99) and nitrogen elimination following E–Z photoisomerization of aliphatic azo compounds (Scheme 6.166). The Norrish type I reaction proceeds preferentially from the n,p states of ketones. State correlation diagrams,331 tunnelling calculations, ab initio and DFT calculations have been used to interpret the relative reactivity of n,p and p,p singlet and triplet states, the rates and the preferred sites for s- bond homolysis (for further references, see ref. 423). The reactivities of singlet and triplet n,p -excited ketones are comparable, but ISC to the triplet state is very fast in aryl ketones (1011 s 1 versus 108 s 1 in saturated ketones)
The rate constants for the reaction of carbon-centred radicals with various substrates such as alkenes424,425 and dioxygen426,427 vary over many orders of magnitude depending
on thermodynamic, steric and stereoelectronic effects. Radical recombination reaction rate constants krec are often close to the diffusion-controlled limit, kr ¼ kd/4;58 the factor of one-quarter is due to spin statistics (Section 2.2.1). The observed second-order rate constant for self-termination reactions is 2kr.
The overall multiplicity of geminate radical pairs formed by bond fission is the same as that of the precursor excited state. Remarkably, the multiplicity of the precursor can often be established by NMR spectroscopy of the final products thanks to a phenomenon called chemically induced dynamic nuclear polarization (CIDNP, Special Topic 5.3).
Special Topic 5.3: Chemically induced dynamic nuclear polarization
The discovery of CIDNP was marked by the serendipitous observation of emission (negative peaks) in NMR spectra of reacting systems.428,429 A personal view of the
early investigations of the CIDNP phenomenon has appeared.430
CIDNP is based on the following principle:431,432 Initially, the radical pair is born in a spin-correlated state. To form a product in the singlet ground state, the electronic spin state of the radical pair must be a singlet state. Importantly, the electron spins interact with the nuclear spin states. ISC from a triplet to a singlet radical pair is favoured, when

Primary Photochemical Intermediates: Examples and Concepts |
205 |
the total (nuclear plus electronic) spin is conserved. Therefore, certain nuclear spin states will favour the recombination of triplet pairs. Initially, the nuclear spin states are at Boltzmann equilibrium.b However, if reaction pathways starting from an overall triplet pair yield different products than those of singlet pairs, CIDNP is observed. The nuclear spin states are unevenly distributed to the different products and, as a consequence, its NMR signals appear either in enhanced absorption or emission.
The reaction between two spin-correlated geminate radicals leading to the final products is called a cage reaction. When the lifetime of a triplet pair is insufficient to permit ISC to the singlet state, the two radicals separate and these free radicals eventually find new reaction partners; this is termed an escape reaction. Each of these reaction types leads to differently polarized NMR signals, that is, the resonances appear in enhanced absorption (A) or emission (E). Kaptein and Closs have developed simple rules, which allow one to determine the multiplicity of the precursor radical pair from the sign of the individual CIDNP signals (A or E) and magnetic parameters of the shortlived radical pair (isotropic hyperfine coupling constants and g-factors).433
bThe energy differences between nuclear spin states are very small compared with kBT even in strong magnetic fields, so that in thermal equilibrium all nuclear spin states are populated nearly equally in accord with Boltzmann’s law (Equation 2.9). This is the reason why NMR spectroscopy, which displays the transitions between different nuclear spin states, is relatively insensitive: One needs milligram quantities for a good spectrum, whereas a few micrograms are usually sufficient to record a UV spectrum. Electromagnetic radiation of the proper frequency produces both absorption and stimulated emission (Figure 2.4) from the sample, and the net absorption is due to the tiny excess of molecules in the lower spin state.
Remarkable effects of nuclear spin on the photochemistry of dibenzyl ketone and its derivatives (see Special Topic 6.11) have been reported and substantial isotopic enrichment of 13C in specific products has been achieved under favourable conditions.434 The influence of magnetic fields and of microscopic reactors (micelles, zeolites, etc.) on these reactions was investigated in detail.435
The radical ion pairs generated by photoinduced electron transfer (Section 5.2) have several options for further reaction.341,365,436 They can undergo return electron transfer
(RET) to regenerate the parent systems in the ground state. Due to the near-degeneracy of the overall singlet and triplet state of radical pairs, ISC in the radical pair may be much faster than in the parent aromatic compounds and RET then leads to the formation of one
of the reactants in the triplet state within 1 ns ( fast triplet formation). ISC in radical ion pairs been attributed to hyperfine coupling on the basis of magnetic field effects.437,438 The
radical cations of strained systems can rearrange, because the barriers to rearrangement are frequently much lower in the transient radical ions than in the parent systems (a wellknown example is the rearrangement of quadricyclane to norbornadiene, Scheme 6.139). The observation of CIDNP signals (Special Topic 5.3) has proven to be a powerful tool to assess the mechanism of such reactions.
The radical cation of naphthalene has been generated in aqueous acetonitrile both by twophoton ionization and via reaction with SO4. . Its lifetime in aqueous solution is 25 ms.439 The equivalent to the triplet state of molecules with a singlet ground state is the quartet state (S ¼ 3/2) in radicals. Quartet configurations should, according to Hund s rule, have a lower energy than the corresponding excited doublet configurations with three unpaired electrons. However, such configurations with three unpaired electrons are generally of