

Photochemistry_of_Organic
.pdf206 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
|||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
radical cation |
radical anion |
neutral radical radical cation with |
|
|
a degenerate |
|
|
ground state |
Figure 5.12 Quartet states of radicals. Single electron excitations leading to a configuration with a single unpaired electron are marked with a full arrow, those leading to a configuration with three unpaired electrons with a dashed arrow
higher energy than the lowest excited doublet configuration, as indicated by the first three MO schemes on the left of Figure 5.12. Thus ISC to a long-lived quartet state is possible only from upper excited doublet states of radicals, which is unlikely to happen because internal conversion within the doublet manifold will usually be much faster. Therefore, quartet ! doublet phosphorescence is difficult to observe and the quartet states of radicals rarely play a role in photochemistry.
The situation is different for radical ions in which the unpaired electron is situated in a degenerate (or nearly degenerate) MO (Figure 5.12, right). Here, the doublet ground state of the radical ion will be degenerate (or nearly degenerate) and the lowest excited state may be one with three unpaired electrons. Indeed, quartet states and quartet ! doublet
phosphorescence of such systems have been observed, for example, for the decacyclene radical anion,440,441 or for the radical cations of acetylene and benzene.442,443
5.4.4Biradicals
Recommended reviews and textbooks.16,318,444
This section is intended to provide the organic chemist with relatively simple guidelines, rational structure–reactivity relationships and rules-of-thumb to predict the reactivity of biradicals and their response to changes in manageable parameters such as temperature, solvent polarity and magnetic fields. The same considerations hold, mutatis mutandis, for carbenes and nitrenes.
The energy of near-UV photons is on the order of bond energies in organic molecules. Photoreactions are thus frequently initiated by the homolytic cleavage of a covalent bond, which leads to a biradical, if the primary product is still covalently linked (Figure 5.13). Biradicals, also called diradicals, are usually defined as molecular species that possess two (possibly delocalized) radical centres, that is, one bond less than the number predicted by the standard rules of valence. When the two radical centres are located on the same atom (1,1-biradicals), such hypovalent species are referred to as carbenes (Section 5.4.1), nitrenes (Section 5.4.2), and so on. The two radical centres may be connected through saturated covalent bonds (localized biradicals) or they may be delocalized over the same p-system (conjugated biradicals).
Chemical transformation is about breaking bonds and forming new bonds. Biradicals are at this interface, so their properties are of fundamental interest. Molecular structures representing biradicals correspond to the funnels connecting excitedand ground-state potential energy surfaces (Figure 2.26). Owing to the near degeneracy of several electronic

|
Primary Photochemical Intermediates: Examples and Concepts |
207 |
|||
|
MO |
|
|
VB |
|
2 e |
? |
|
|
|
|
Pauli |
|
|
|
|
|
|
|
|
|
|
|
NBMOs |
• |
• |
|
|
|
|
|
|
|
||
|
3 singlet states |
1 covalent singlet state |
2 zwitterionic |
|
|
|
1 triplet state |
(dot–dot) |
|
(hole–pair) |
|
|
|
1 triplet state |
|
singlet states |
|
Figure 5.13 The four low-lying electronic states of a biradical
states, the electronic structure of biradicals is chameleon-like, which complicates anticipation of their chemical properties, but also opens up the opportunity of influencing their behaviour. Their intrinsic instability has long discouraged attempts to exploit their synthetic potential as bifunctional reagents. Systematic trapping studies, the spectroscopic detection and identification of biradical intermediates in solid matrices and in solution, investigations of their reaction kinetics in solution, combined with the development of theoretical models, have brought some clarification and led to the identification of relatively long-lived biradical intermediates that can be of interest for synthesis.
A molecule with two radical centres will have two nonbonding MOs that are occupied by a total of two electrons; if these electrons have parallel spin, they must occupy one NBMO each, forming a triplet configuration. If they have opposite spin, two additional configurations can be constructed, in accord with the Pauli principle, by placing both electrons into one or the other NBMO. We obtain one triplet state and three singlet states (Figure 5.13), all of which have the same energy in terms of HMO theory. When we consider electronic interaction, the triplet state is likely to be the ground state of the biradical (Hund s first rule) and the two singlet states with localized charges (hole–pair states), represented as zwitterions in a valence bond (VB) description, are usually pushed higher in energy than the so-called covalent singlet state with one electron on each radical centre.
The definition of a biradical given above is based on G. N. Lewis s rules of valence and would exclude molecules such as cyclobuta-1,3-diene (3), which have a structure satisfying these rules, that is, a Kekule structure. Yet 3 has two NBMOs according to HMO theory (Section 4.2, Figure 4.2) and therefore exhibits the electronic properties of a biradical. One might then define a biradical as an open-shell species with two electrons in two (essentially) nonbonding orbitals. Instead, we prefer the following practical definition of a biradical, which avoids reference to any model (VB or MO): a biradical is a molecular species having three low-lying electronic singlet states and a triplet state with an energy either below or little above the lowest singlet state.445 Because the thermal population of low-lying electronic states depends on temperature, the singlet–triplet energy gap should be judged relative to kBT (NAkBT ¼ RT ¼ 2.5 kJ mol 1 at room temperature). Frequently, the NBMOs are split apart to some extent, for example when the two radical centres interact substantially, when they have intrinsically different electronegativity or when structural relaxation, such as bond localization in cyclobutadiene, stabilizes one NBMO relative to the other. Biradical-like species with a singlet–triplet energy gap exceeding kBT may be referred to as biradicaloids.
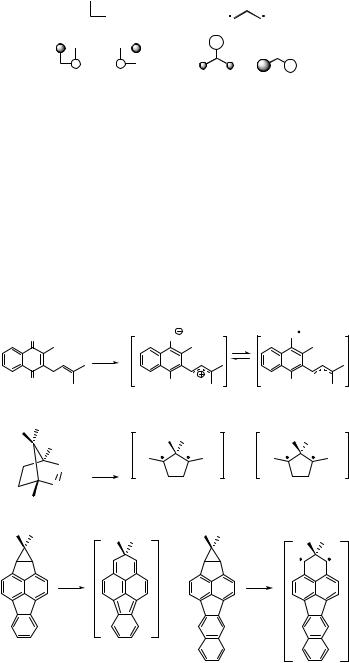
A distinction is made between conjugated Kekule biradicals such as 3, for which at least one Kekule structure can be drawn, and non-Kekule biradicals such as
208 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||||||||||
|
|
|
o |
|
|
|
* |
|
|
|
|
* |
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
o |
|||
|
* |
|
|
|
o |
* |
|
||||||
|
|
|
|
* |
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
cyclobutadiene 3 trimethylenemethane 17
Figure 5.14 NBMOs of cyclobutadiene and trimethylenemethane
trimethylenemethane (17), which does not have a proper Kekule structure (Figure 5.14). In non-Kekule alternant hydrocarbons, the number of starred ( ) atoms s exceeds that of the unstarred (o) atoms u, so that the number of NBMOs must be at least equal to |s – u|, that is, 2 (Section 4.6). Hence all non-Kekule hydrocarbons are biradicals. For Kekule hydrocarbons, |s – u| 0; they are normally stable molecules with a closed-shell ground state such as benzene or butadiene. However, some Kekule hydrocarbons such as cyclobutadiene (3) have two NBMOs or a very small energy gap between the HOMO and LUMO; they are called Kekule biradicals or biradicaloids. Two other Kekule biradicals (20 and 21) are shown in Scheme 5.10.
Another important classification of conjugated diradicals depends on whether their H€uckel NBMOs can or cannot be confined to a disjoint sets of atoms.446 As discussed in
|
O |
|
1 |
O |
3 |
O |
|
|
hν |
|
|
|
|
|
O |
|
|
OH |
|
• |
|
|
|
|
OH |
||
|
18 |
|
λmax = 1400 nm |
λmax = 660 nm |
||
X |
X |
|
1 |
RO OR |
3 |
Me Me |
|
|
|||||
|
Ph |
|
||||
|
|
|
|
|||
|
|
|
Ph |
|
Ph |
|
|
N |
hν |
Ph |
Ph |
||
|
|
|
|
|
||
|
N |
–N2 |
λmax = 570 nm |
λmax = 440 nm |
||
|
Ph |
|
||||
19, X = OR or Me |
|
|
|
|
|
|
|
1 |
|
|
|
|
3 |
|
hν |
|
|
|
hν |
|
22 |
20 |
|
|
|
λmax = 1400 nm |
23 |
21 |
λmax = 670 nm
Scheme 5.10 First absorption bands of singlet and triplet biradicals
Primary Photochemical Intermediates: Examples and Concepts |
209 |
Section 4.2, any normalized linear combination of degenerate wavefunctions is an equally valid wavefunction. The linear combinations of the NBMOs with the smallest possible amount of local overlap for 3 and 17 are shown in Figure 5.14. Those of 3 are separated entirely in space to the starred and unstarred sets of atoms. Hence 3 is a disjoint biradical. The same cannot be achieved by any linear combination of the NBMOs of 17; it is classified as a nondisjoint biradical.
The distinction between disjoint and nondisjoint conjugated biradicals provides a useful basis for understanding their electronic properties. The class to which a given biradical belongs can be immediately assessed by PMO analysis (Section 4.6). The union of an allyl radical with a methyl radical to form either 3 or 17 will give two NBMOs in either case, because it takes place at a nodal plane of the NBMO of the allyl radical. The NBMO coefficients are then easily obtained from rule 4 (Equation 4.25): the coefficients on atoms attached to an atom with cNBMO,m ¼ 0 must add up to zero. PMO analysis directly gives the localized NBMOs, which keep the electrons in the open shell apart as much as possible and which are best suited to describe the lowest energy singlet and triplet states by a single configuration.
Starting from this localized basis set, the singlet–triplet gap DEST ¼ ES – ET can be estimated as two times the exchange integral Kij (Equation 4.34) between the two NBMOs ci and cj, which will be substantial (on the order of 1 eV) for nondisjoint biradicals such as
17, but vanishes within the ZDO approximation for disjoint biradicals such as 3. The triplet state is indeed the ground state of 17 and the DEST amounts to 67.4 0.4 kJ mol 1.447Ab
initio calculations predict that the equilibrium geometry of 317 is planar (D3h symmetry), whereas 117 relaxes to a perpendicular geometry in which one methylene group is orthogonal to the plane of the other atoms (C2v).448 On the other hand, cyclobutadiene (3) has a singlet ground state that is stabilized by bond length alternation. Our simple model gives DEST(3) ¼ 0, but ab initio calculations slightly favour the singlet even for planar quadratic 3 due to a small stabilization called spin polarization. Allowing for this minor correction, the classification based on the simple PMO procedure provides robust predictions for the ground state multiplicity of many conjugated biradicals.
The exchange integral falls off rapidly with distance (Figure 4.21) and so does the singlet–triplet gap with increasing separation of the radical centres. In 1,2-biradicals (90 twisted alkenes, Section 5.5) and in localized 1,n-biradicals (n > 2) the gap is usually within a few kJ mol 1, that is, comparable to RT at room temperature. The energy gap DEST plays a decisive role in ISC processes of biradicals.
Because biradicals have three singlet states of low energy, one may predict in general that singlet biradicals will absorb at longer wavelengths than triplet biradicals of the same (or similar) structure. This is illustrated by the MO diagram in Figure 5.15, which shows the two additional low-energy transitions available to singlet biradicals.
Dioxygen is a typical example (Section 2.2.5). In the triplet ground state it absorbs only in the deep UV region, but the lowest excited state of singlet oxygen (1Sgþ ) lies only 62.7 kJ mol 1 above the lowest singlet state (1Dg). Other examples of such longwavelength absorption bands in organic singlet biradicals are shown in Scheme 5.10.
The top row of Scheme 5.10 shows the m-quinone methide biradical that is formed by photolysis of menaquinone-1 (18), an analogue of vitamin K1.449 The singlet and triplet state of this biradical are in thermal equilibrium. The zwitterionic singlet is stabilized in polar media and absorbs at 1400 nm. The second row shows 2,2-disubstituted 1,3-

210 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
triplet singlet
Figure 5.15 MO scheme of electronic transitions in singlet and triplet biradicals
diphenylcyclopentane-1,3-diyl biradicals that are generated by photolysis of the corresponding azoalkane precursors 19. The 2,2-dimethyl derivatives have a triplet ground state and their absorption spectra are similar to those of benzyl radicals, showing a moderately strong band at 320 nm and a very weak, parity-forbidden band at 440 nm.450 The singlet state is the ground state in the 2,2-difluoro- and 2,2-dialkoxy-substituted derivatives, which show a broad, strong absorption band in the visible region, lmax ¼ 500–600 nm.451 The bottom row shows two nonalternant hydrocarbons, 2,2-dimethyl-2H-benzo[cd] fluoranthene (20) and its benzannellated analogue 2,2-dimethyl-2H-dibenzo[cd,k]fluor- anthene (21), that were formed by photolysis of their cyclopropane precursors 22 and 23, respectively.452 Compound 21 is the only known Kekule biradical with a triplet ground state. The singlet–triplet gap for 20, which has a singlet ground state, was estimated to be10 kJ mol 1 and that of 21 (triplet ground state) about þ 5 kJ mol 1. Although 321 is more extended, its first absorption band lies at much shorter wavelength than that of 120. The
saturated singlet carbene adamantylidene exhibits a broad, weak absorption band at 620 nm.398
Scheme 5.11 shows a general blueprint that serves as a useful framework for considering reaction mechanisms that involve biradical intermediates B. The scheme also applies to carbenes (Section 5.4.1) and nitrenes (Section 5.4.2). When formed by thermal or photochemical homolytic bond cleavage from the reactant molecule M, biradicals will initially have the multiplicity of their immediate precursors. Singlet biradicals (1B) tend to be short-lived because spin-allowed bond formation leading back to the reactant M or to a
1M* |
ISC |
|
|
3M* |
|
|
|
|
3B* |
||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
hν |
|
hν |
|
sens |
|
1B |
|
|
3O2 |
|
|
||||
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
3B |
products (M'') |
||||
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
3O |
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
∆ |
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3O |
||
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
peroxides M–O2 |
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
||||||
M |
|
products (M') |
|
|
dimers |
||||||||
Scheme 5.11 A general reaction scheme for reactions via biradical intermediates
Primary Photochemical Intermediates: Examples and Concepts |
211 |
||||
S |
ISC |
T |
|
3B* |
|
1 |
|
1 |
|
|
|
|
|
|
–N2 |
hν |
|
|
–N2 |
|
|
|
|
|
|
|
|
|
|
hν |
sens |
|
3O2 |
|
|
|
|
|
∆ |
|
|
|
|
124 |
324 |
|
|
|
∆ |
–N |
dimerization |
3O2 |
|
|
|
2 |
|
|
|
N |
N |
|
|
O O |
|
+
2
Scheme 5.12 Singletand triplet-state reactivity of a conjugated hydrocarbon biradical453
new product is generally fast (picosecond time scale). ISC to the triplet state of the biradical (3B) is unlikely to compete in hydrocarbon biradicals, except in carbenes (Section 5.4.1) or at low temperatures, if bond formation requires some activation energy.
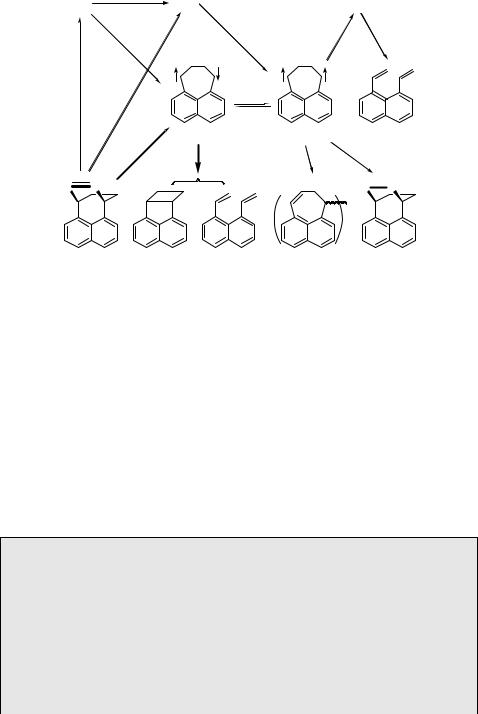
The triplet biradicals may be generated from the triplet state of the reactant M. They are generally longer lived (nanoseconds) because they need to undergo ISC to return to a singlet ground-state product. In aerated solution, they can be trapped by dioxygen to form peroxides (or carbonyl compounds in the case of carbenes). Similarly, dimers of 3B may be formed under high-intensity irradiation. These are spin-allowed processes for one in nine encounters (Scheme 2.5). At low temperature, triplet biradicals may be persistent and can be identified by ESR spectroscopy. Light absorption by 3B in flash photolysis or at low temperature may lead to new products via an excited-state 3B . Practical examples are provided by the photoreactions of the azo precursor of 1,4-perinaphthadiyl (24)453, Scheme 5.12, and Case Study 5.2.
Case Study 5.2: Mechanistic photochemistry – cyclopentane-1,3-diyl biradicals
1,3-Diphenylclopentane-1,3-diyl454,455 (Scheme 5.13) can be generated in its singlet state by either direct irradiation or thermolysis of its azo precursor and also by thermal homolysis of the central bond of 1,4-diphenylbicyclo[2.1.0]pentane (housane). Homolysis of this bond releases a large amount of strain energy. Moreover, the resulting biradical is stabilized by the phenyl groups. Thus the central bond of housane is extraordinarily weak and the degenerate ring-flip reaction, which must proceed via the CP biradical either as the transition state or as an intermediate, occurs about twice per
212 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||||
|
|
|
|
Ph |
|
|
|
|
|
|
|
N |
|
|
|
|
|
|
|
N |
|
|
|
|
|
∆or hν |
|
Ph |
|
|
|
|
|
|
|
|
sens. |
|
|
|
Ph |
|
Ph |
3O2 |
Ph |
Ph |
|
|
|
|
|
||||
|
∆ |
1CP |
∆ |
|
|
3CP 3O2 |
Ph |
|
Ph |
|
|
Ph |
|
|
O |
|
|
|
Ph |
|
|
O |
|
|
Ph |
|
|
|
Ph |
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
Scheme 5.13 |
|
|
||
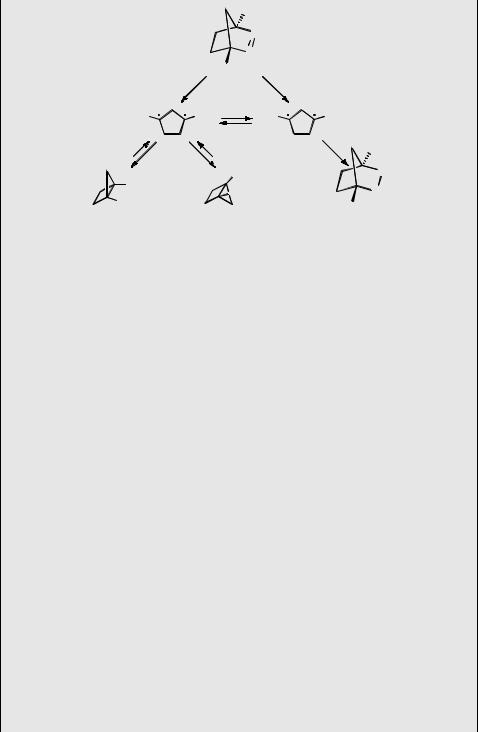
second at ambient temperature. The activation parameters for ring-flip were determined by H-NMR as DHz ¼ 51.0 0.4 kJ mol 1 and DSz ¼ 69 6 J K 1 mol 1.455
Does 1CP lie in a potential well and does it undergo spontaneous ISC to 3CP? The answer to the second question is no.454 This was established by flash photolysis and by oxygen trapping. The triplet state 3CP was identified by its ESR spectrum, which persists down to 3.8 K, indicating that the triplet state is the ground state of the biradical. Flash photolysis of benzophenone to sensitize the diazo precursor forms 3CP, which has a lifetime of 27 ms (lmax ¼ 320 nm) in degassed solution. Oxygen quenching of 3CP is fast, k ¼ 7.5 109 M 1 s 1, and yields the endoperoxide. Upon direct flash photolysis of the azo compound, 3CP is not observed and the housane is formed in high yield. The latter is stable in degassed solution, but it reacts with oxygen forming the endoperoxide in aerated acetonitrile. The rate law for oxidation is first order in both oxygen and housane concentration, kox ¼ 2.1 M 1 s 1 at 24 C. Thus, at an oxygen concentration of 2 10 3 M (air-saturated) or less, the ring-flip reaction, kflip ¼ 2 s 1, is much faster than that of oxidation, kox[O2] ¼ 4.3 10 3 s 1. These observations exclude oxygenation occurring by spontaneous ISC of 1CP followed by trapping of 3CP, because any 3CP formed would be quantitatively trapped and the rate law would be zero order in oxygen concentration.
Returning to the first question, the answer is yes. 1CP is a metastable intermediate, as is already indicated by the fact that oxygen trapping does occur. Any encounter of 1CP with 3O2 will induce oxygen-catalysed ISC, a spin-allowed process (Section 2.2.5). Once formed, 3CP is likely to leave the encounter complex with 3O2, which has overall triplet multiplicity and cannot form the endoperoxide directly. However, formation of 3CP is irreversible, as 3CP will rapidly be trapped by another oxygen molecule. Assuming from many precedents that catalysed ISC occurs at the diffusion limit,
k |
10 |
10 M 1 |
|
1 |
|
1 cat 2.8 |
|
s |
|
, the observed oxidation kinetics indicates that the lifetime of |
|
CP is at least 20 ps and that the barrier protecting it from cyclization amounts to about |
|||||
12 kJ mol 1.454 These results are illustrated by the PES for the ring-flip reaction shown in Figure 5.16.

Primary Photochemical Intermediates: Examples and Concepts |
213 |
E |
|
T1 |
S0 |
|
|
|
SOC |
|
|
|
Ph |
Ph |
Ph |
Ph |
Ph |
Ph |
|
|
|
|
|
|
Figure 5.16
How does a triplet biradical 3CP return to the singlet ground state? As shown in Figure 5.16, it will have to pass a small barrier to the intersection with the singlet PES and the magnitude of spin–orbit coupling (SOC) (Section 4.8) at the crossing point will determine the small probability for diabatic reaction (Landau–Zener model, Equation 2.53), that is, that the molecule will find the escape hatch to the singlet ground state. Application of a general analysis of SOC terms in biradicals320 to the case of 3CP shows that SOC of the triplet state to the lowest singlet state is zero for a planar 3CP biradical, because the lowest singlet state wavefunction (covalent, dot–dot) cannot mix with the zwitterionic wavefunctions (hole–pair) that are needed to induce SOC with the covalent triplet state (Section 4.8). This already explains the remarkably long lifetimes of the geometrically constrained 3CP biradicals. However, these lifetimes are fairly sensitive to the structural environment, as indicated by the examples shown in Scheme 5.14. Clearly, 2,2-dimethyl substitution strongly reduces the lifetimes of these geometrically constrained triplet 1,3-biradicals.
To understand this trend, we need to look at the requirements permitting SOC through admixture of the lower zwitterionic (hole–pair) state to the covalent singlet ground state wavefunction. Two conditions must be met:320 the molecule must bend
|
Ph |
Ph |
|
260 ns |
16 s |
>20 ms |
59 ns |
|
Ph |
Ph |
|
< 0.1 ns (?) |
1.3 s |
400 s |
4–14 ns |
Scheme 5.14 Lifetimes of some triplet 1,3-biradicals in degassed solution450,456–459

214 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||
|
|
through |
through |
|
|
|
|
space |
bond |
|
H |
|
|
|
a |
|
|
|
|
|
|
H |
|
|
H |
a |
γ |
|
|
|
|
|
|||
|
H |
|
H |
||
|
|
|
|
||
|
|
|
s |
|
H |
|
H |
s |
|
|
|
|
H |
|
|
|
|
|
|
|
|
s |
H |
|
|
|
|
H |
|
|
|
H |
|
|
|
|
|
|
|
pseudo- |
|
|
|
|
|
|
|
|
|
H |
|
|
|
Figure 5.17 MO interaction diagram for cyclopentane-1,3-diyl (CP). The symmetry labels a and s refer to reflection through the plane defined by the CH2 group at position 2
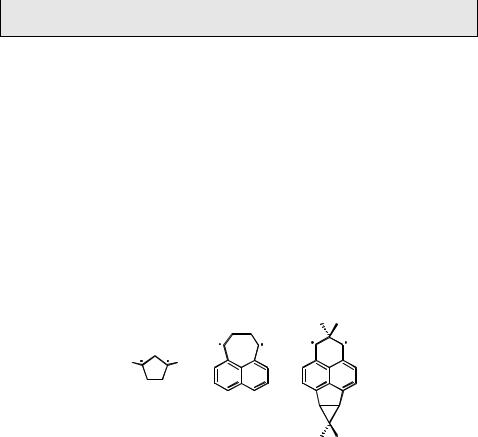
along the reaction coordinate leading to housane (Figure 5.16) and the two NBMOs must be split by a covalent interaction. Consider the simplified MO diagram for planar CP shown in Figure 5.17.
The interaction between the two symmetry-adapted NBMOs s (symmetric with respect to the plane of the CH2 group at position 2) and a (antisymmetric) shown on the left is considered in two stages. The direct through-space interaction stabilizes the s and destabilizes the a NBMO. This is counteracted by through-bond interaction involving the pseudo-p MO of the 2-CH2 moiety, resulting in a small energy gap g separating the two NBMOs, which house one electron each in the triplet state of the biradical. The matrix element that induces mixing of the lower-lying, closed-shell zwitterionic (hole–pair) wavefunction with the covalent wavefunction is g. Hence the rate constant of ISC will be proportional to g2.450 By replacing the CH2 group at position 2 with a CMe2 group, the basis energy of the pseudo-p orbital interacting with the s- combination of the p-NBMO will be raised by about 1.5 eV. As a result, the symmetric NBMO is raised well above the antisymmetric one and the energy gap g is increased. This will increase the contribution of the hole–pair wavefunction to S0 at nonplanar geometries leading to increased rates of ISC in 2,2-dimethyl-substituted CP biradicals.457
Another modification that has been introduced into the parent 1,3-diphenyl CP biradicals is substitution at the phenyl groups. A series of 33 symmetrically and asymmetrically substituted triplet 1,3-diaryl CPs was investigated by flash photolysis.450 Arrhenius parameters were determined from the temperature-dependence of the ISC rate constants, yielding activation energies Ea in the range 8–25 kJ mol 1 and preexponential factors A of 107–1010 s 1. The low A-factors reflect the low probability of ISC at the surface crossing. Counter to intuition, but in agreement with the model,320 asymmetric push–pull substitution does not enhance the contribution of the hole–pair wavefunctions to S0 and, accordingly, does not enhance ISC rates. The observed rate constants correlate well with calculated values of g2, except for bromoand iodo-
Primary Photochemical Intermediates: Examples and Concepts |
215 |
substituted compounds, which exhibit enhanced ISC rates attributable to the heavy-atom effect (Section 4.8).
Several singlet biradicals were generated by femtosecond-pulsed decarbonylation of cyclic ketones and their femtosecond dynamics were investigated by time-resolved mass spectrometry.460 Very short lifetimes were reported for singlet trimethylene (.CH2CH2CH2., 120 fs), tetramethylene (.CH2CH2CH2CH2., 700 fs) and parent CP (190 fs). It should be noted, however, that these biradicals were formed with substantial excess energies as isolated species in the gas phase (the CP biradical was formed by absorption of two 307 nm photons). Several singlet 1,4-biradicals were formed by the Norrish type II reaction (Section 6.3.4) of pentan-2-one and their lifetimes in the range 0.5–0.7 ps were determined by the same method.461 The lifetimes of these biradicals, in particular that of CP, might be substantially higher in solution. A few singlet biradical lifetimes have been estimated by oxygen trapping, for example 1,3-diphenyl-CP (Case Study 5.2) and 1,4-perinaphthadiyl (24, Scheme 5.12),453 or measured by pump–probe spectroscopy [the 1,3-biradical intermediate in the photocyclization of 2,7-dihydro- 2,2,7,7-tetramethylpyrene, Figure 6.11], Scheme 5.15.
Ph |
Ph |
|
|
1,3-diphenylCP |
24 |
|
|
20 ps (O2-trapping) |
1 ns (O2-trapping) |
18 ps |
|
Scheme 5.15 Singlet biradical lifetimes
A series of localized triplet 1,n-biradicals was generated by laser flash photolysis of the cyclic ketones shown in Scheme 5.16 (Norrish type I cleavage, Section 6.3.3).40 For small chain lengths, the main products were formed by disproportionation, but for n 10, paracyclophanes were obtained in high yield. The lifetimes of the triplet biradical intermediates (lmax ¼ 320 nm) increased with chain length from 45 ns (n ¼ 6) to 80 ns (n ¼ 8) and then decreased again to 50 ns (n ¼ 12 and 15). ISC to the singlet state is thought to be the rate-determining step in their product forming reactions. Both the singlet–triplet energy gap DEST and SOC fall off exponentially with the distance between the radical centres. ISC rates will be different for each conformer of these flexible biradicals and subsequent product formation occurs from a small subset of singlet conformers having a short end-to-end distance. Even though the biradicals will spend most of their time in extended conformations, ISC induced by SOC will take place from a few conformers with a relatively small distance separating the radical centres. Such conformations are sparse for n ¼ 8 and these biradicals have the longest lifetime.