

Photochemistry_of_Organic
.pdf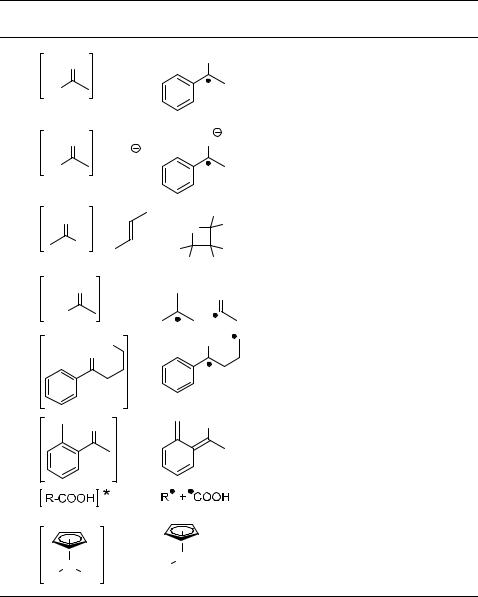
Oxygen Compounds |
297 |
Table 6.6 Examples of photoprocesses involving excited oxygen-atom containing compounds.
Entry Starting |
|
Product(s) |
Mechanism |
Section |
|
materiala |
|
|
|
|
|
O |
* |
|
OH |
Intermolecular |
|
|
|
|
|||
1 |
+ |
[H] |
|
H-transfer |
6.3.1; 6.3.7 |
Ph |
|
|
|
(photoreduction) |
|
O |
* |
|
O |
Intermolecular |
|
2 |
+ |
[e ] |
|
e-transfer |
6.3.1; 6.3.7 |
Ph |
|
|
|
(photoreduction) |
|
O |
* |
|
H |
[2 þ 2] cycloaddition |
|
O |
CH |
|
|||
3 |
+ |
3 |
(oxetane formation; |
6.3.2; 6.3.7 |
|
H |
|
Me |
CH3 |
Paterno`–Buchi€ reaction) |
|
|
|
|
|
||
|
|
H |
H |
|
|
O |
* |
|
O |
a-Cleavage |
6.3.3 |
4 |
|
|
|||
|
+ |
|
|||
t-Bu |
|
|
(Norrish type I reaction) |
|
|
|
|
|
|
||
|
H |
* |
OH |
|
|
|
|
|
|
||
5 |
O |
|
|
Intramolecular |
|
|
|
|
|
||
|
|
|
|
H-transfer (Norrish type II |
6.3.4; 6.3.5 |
|
|
|
|
reaction) |
|
|
O |
* |
OH |
|
|
|
|
|
|
||
6 |
|
|
|
Photoenolization |
6.3.6 |
7 |
|
|
|
Photofragmentation |
6.3.8 |
8 |
* |
|
+ CO |
Photodecarbonylation |
6.3.9 |
|
R M |
||||
M |
|
|
|
|
|
R CO |
|
|
|
|
|
a[H] ¼ hydrogen atom donor; [e ] ¼ electron donor.
Hydrogen abstraction (Section 4.9) by excited carbonyl compounds is one of the most fundamental reactions in organic photochemistry.862–864 In a classical871 photoreduction reaction, an excited carbonyl compound, such as a ketone, undergoes hydrogen abstraction from a hydrogen donor [H] to form the ketyl radicals, which subsequently abstract another
298 |
|
|
|
Chemistry of Excited Molecules |
|
|
|
|
|
|||||||||
|
|
|
|
|
|
1 |
|
|
|
3 |
|
|
|
|
|
* |
||
|
O |
|
|
O |
|
O |
|
|||||||||||
|
|
hν |
|
* ISC |
|
|
||||||||||||
|
R1 |
|
R2 |
|
|
R1 |
|
R2 |
R1 |
|
|
|
R2 |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
hydrogen |
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
[H] |
abstraction |
|
|
|
[H] |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
OH
|
OH |
|
R1 |
R2 |
|
coupling |
|
|
|
|
|
|
|
|
1 |
2 |
reaction |
[H] |
hydrogen |
R |
R |
|
abstraction |
|
|
|
|
|
HO |
OH |
|
|
OH |
||||||
R1 |
|
|
|
|
R1 |
R1 |
|
|
H |
|
|
|
|
|
|
|
|||||
|
R2 |
R2 |
|
R2 |
||||||
256
Scheme 6.99
hydrogen atom from the environment or recombine to form either alcohols or diols (pinacols, 256) (Scheme 6.99).
Photoreduction of aliphatic ketones may involve both singlet and triplet excited states, but the quantum yield for product formation via singlet is usually low because other competing processes, such as radical pair recombination, are involved. The rapid intersystem crossing (ISC) in aryl ketones (Section 2.1.6) allows triplet reactivity. Ketones with n,p lowest triplets, having an unpaired electron localized in an n-orbital on oxygen, are far more reactive than those with p,p lowest triplets.863
The rate constants of the hydrogen abstraction by triplet n,p excited carbonyl compounds are strongly dependent on the reaction thermodynamics. (1) The higher the excitation energy of the hydrogen acceptor, the faster is the reaction. For example, triplet excited a-diketones,872 such as biacetyl (ET ¼ 236 kJ mol 1), will generally undergo much slower (less exothermic) hydrogen abstraction than phenyl ketones (ET 300 kJ mol 1).
(2) Correspondingly, low dissociation energies of the H S bond (DS H) in the hydrogenatom donors favour the abstraction, which parallels the energetics of hydrogen abstraction by simple alkoxy radicals. 873 For example, benzophenone triplet (ET ¼ 288 kJ mol 1) will abstract hydrogen from the secondary C–H bond of propan-2-ol (DC H ¼ 381 kJ mol 1) more rapidly than from the corresponding O H bond (DO H ¼ 442 kJ mol 1) or the C–H bond of neopentane (DC H ¼ 420 kJ mol 1), but more slowly than from tributylstannane (DSn H ¼ 326 kJ mol 1) (Table 6.7).
Considerable photoreductive reactivity is also observed for substrates which are poor hydrogen donors but good electron donors (i.e. possessing a low Ei) (Table 6.7). Such compounds are able to reduce efficiently both p,p and n,p triplets via partial or complete electron transfer (Section 5.2) followed by proton transfer. Amines are very common electron donors; depending on their structure, either an N H or C H bond is cleaved via a
Oxygen Compounds |
299 |
Table 6.7 Rate constants (k) for bimolecular reaction of triplet benzophenone (ET ¼ 288 kJ mol 1) with various substrates (H to be abstracted is shown in bold )
Substrate |
k/106 M 1 s 1 |
DS–Ha/kJ mol 1 |
Ei a/eV |
Neopentane |
0.04 |
420 |
– |
H CH2OH |
0.2 |
402 |
10.9 |
C6H5CH2 H |
0.5 |
370 |
8.8 |
H C(OH)(CH3)2 |
1.9 |
381 |
10.1 |
n-Bu3Sn H |
1300 |
326 |
– |
Ph OH |
1300 |
368 |
8.5 |
Et2N CH2CH3 |
3000 |
377 |
7.3 |
aDS H are bond dissociation energies; Ei are ionization potentials.157,863,874
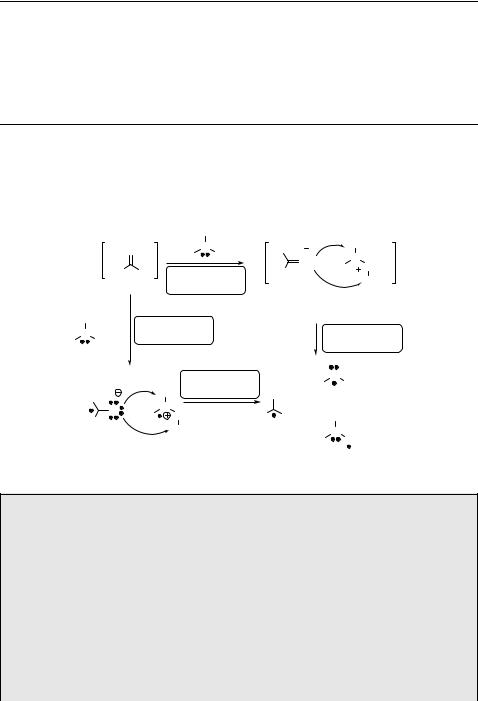
triplet exciplex or an radical ion pair to form the same primary products [Scheme 6.100; pathway (a) or (b), respectively].669,875,876 Reduction of aromatic ketones by amines in
aqueous media usually yields secondary alcohols rather than pinacol derivatives.
|
|
|
H |
|
(a) |
H |
|
|
3 |
O |
* |
N |
R1 δ |
|
|
||
|
|
|
||||||
|
|
R' CH2R'' |
O |
|
N |
|
|
|
R1 |
R2 |
|
|
|
R' δ |
CHR'' |
||
|
charge |
R2 |
|
|||||
|
|
|
transfer |
|
(b) |
|
H |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
H |
|
|
|
triplet exciplex |
|
|
||
|
electron |
|
|
|
|
|
||
N |
|
|
|
hydrogen |
|
|||
|
transfer |
|
|
|
||||
R' CH2R'' |
|
|
|
|
|
transfer |
|
|
|
|
|
proton |
|
|
N |
|
via (a) |
|
(a) |
|
|
R' |
CH2R'' |
|
||
R1 |
H |
transfer |
OH |
|
||||
|
|
or |
|
|
||||
|
|
|
|
|
|
|||
O |
R' |
N |
CHR'' |
+ |
|
|
|
|
R1 R2 |
|
H |
|
|
||||
R2 |
|
|
|
|
||||
|
|
H |
|
|
N |
|
via (b) |
|
|
(b) |
|
|
R' |
|
|||
|
|
|
|
CHR'' |
|
|||
Scheme 6.100
Case Study 6.16: Chemistry in ionic liquids – photoreduction
The amine-mediated photoreduction of benzophenones in ionic liquids at 20 C provides the corresponding benzhydrol derivatives.877 Unlike the analogous reactions in common organic solvents, where pinacol derivatives are predominately formed, 2-methoxycarbonylbenzophenone (257), irradiated in the presence of 2-butylamine in an ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate [EMI(OTf)], gave a nearly quantitative isolated chemical yield of methyl 2-[hydroxy(phenyl)methyl] benzoate (258) (Scheme 6.101). The authors suggest that benzhydrols are produced via a stabilized ion pair, formed from amine and ketone by single electron transfer. Ionic liquids are considered to be environmentally friendly alternatives to conventional solvents.878

300 |
Chemistry of Excited Molecules |
|
O |
COOCH3 |
OH COOCH3 |
|
|
hν |
|
|
NH2 |
257 |
|
, |
|
258 |
|
|
|
ionic liquid |
Scheme 6.101
Experimental details.877 An EMI(OTf) solution of 257 (25 mM) in a closed Pyrex vessel was purged with argon for 1 h and 2-butylamine solution (1 M) was added via a syringe. The reaction mixture was shaken vigorously, irradiated using a mediumpressure Hg lamp (450 W) at lirr > 290 nm (Pyrex optical filter; Figure 3.9) for several hours and the resulting mixture was extracted with diethyl ether. The organic layer was washed with dilute HCl to remove the excess amine. The extracts were dried over MgSO4, concentrated in vacuo and the product was purified by column chromatography.
6.3.2Carbonyl Compounds: Oxetane Formation (Paterno`–Buchi€ Reaction)
O
 +
+
Recommended review articles.584,602,689,751,879–883
Selected theoretical and computational photochemistry references.16,534,884–886
The photochemical [2 þ 2] cycloaddition of an alkene to an excited carbonyl compound, termed the Paterno`–B€uchi reaction,887,888 leads to an oxetane moiety.
Although both singlet and triplet excited species are known to produce cycloadducts, the
scope and regioand stereoselectivity of this process depend on the reactant multiplicity.879,880,882 The reaction may involve a concerted reaction or a stepwise
mechanism via 1,4-biradical intermediates (BR) and the products are formed from either
the C O or C C initial attack producing two different triplet biradicals (Scheme 6.102).879,889 Exciplex, radical ion pair and zwitterion intermediate formation prior to oxetane cyclization have also been noted in some cases.890,891 This transformation
may compete with inter- (Section 6.3.1)/intramolecular hydrogen abstraction (Section 6.3.4) or a-cleavage (Section 6.3.3) of the carbonyl moiety or with the reactions involving the alkene molecule. The course of the reaction then depends on many factors. The carbonyl compound should be the only absorbing chromophore during the irradiation and photostable in the absence of alkene under the given reaction conditions.
The attack of n,p excited carbonyl compounds on electron-rich or electron-poor alkenes can be qualitatively different.892,893 Electron-rich alkenes preferentially interact with the electrophilic half-filled n-orbital on the oxygen atom, which is perpendicular to
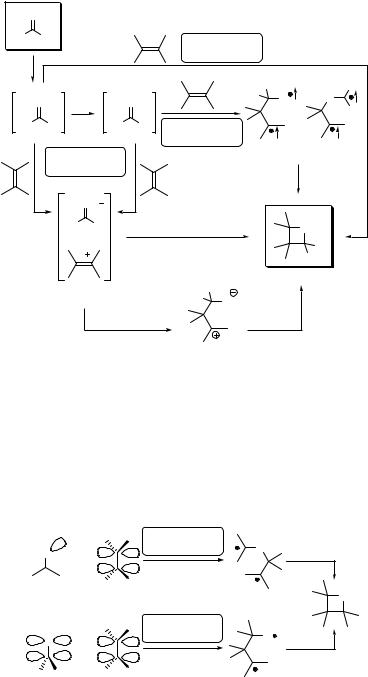

302 |
|
Chemistry of Excited Molecules |
|
|
Table 6.8 Fluorescence quenching of norbornan-2-ones by substituted ethylenes |
|
|||
Ketone |
|
|
kq/(109 M 1 s 1)a |
|
|
|
CN |
EtO |
OEt |
|
|
|
||
|
|
NC |
|
|
|
O |
|
|
|
|
|
5.1 |
|
1.2 |
|
O |
2.3 |
|
0.9 |
|
|
|
||
|
O |
1.0 |
|
1.5 |
|
|
|
||
|
O |
|
<0.03 |
|
|
|
0.5 |
||
aFluorescence quenching rate constants.892
The rate constants and selectivities of the Paterno`–B€uchi reaction are strongly influenced by steric effects.892 Fluorescence quenching (reflecting the first step to oxetane formation) of singlet excited norbornan-2-one derivatives is reduced by increasing the substitution in the vicinity of the carbonyl group (Table 6.8). The rate constants of quenching (kq) by electron-deficient (E)-1,2-dicyanoethylene decrease when the methyl substituents hinder access to the p-plane of the carbonyl group (C-atom attack), whereas those of electron-rich (Z)-1,2-diethoxyethylene are sterically sensitive to the n-plane approach of the carbonyl oxygen (O-atom attack). Interestingly, a similar parallel approach dependence is observed for the ground-state reduction of norbornan-2-ones by NaBH4, which is known to transfer the hydride to the carbonyl carbon atom in the rate-determining reaction step.894
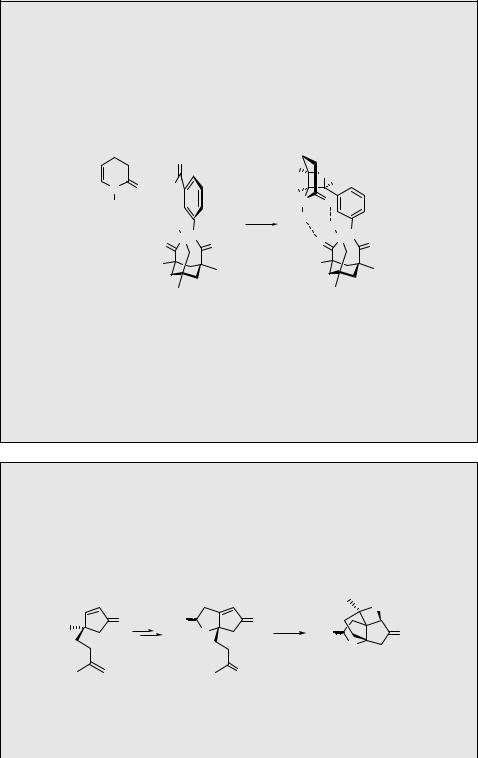
In another example, the reaction selectivity is pronounced by enlarging the ortho substituents of the phenyl ring in triplet excited benzaldehyde, which reacts with dihydrofuran (Scheme 6.104).895 Surprisingly, the cycloaddition produces an excess of the thermodynamically less stable endo-diastereomers, which was explained as formation of specific triplet biradical geometries that undergo rapid intersystem crossing.
The lowest excited singlet states of aliphatic aldehydes and ketones have lifetimes on the order of nanoseconds, but they can be trapped by alkenes in a diffusion-controlled bimolecular oxetane formation. According to a theoretical study, a C-atom attack mechanism is either a concerted process producing oxetane directly or it involves a C C bonded transient singlet biradical intermediate that rapidly cyclizes.896 The O-atom attack, in contrast, represents a nonconcerted path, allowing conformational motion of the shortlived intermediate thereby formed.
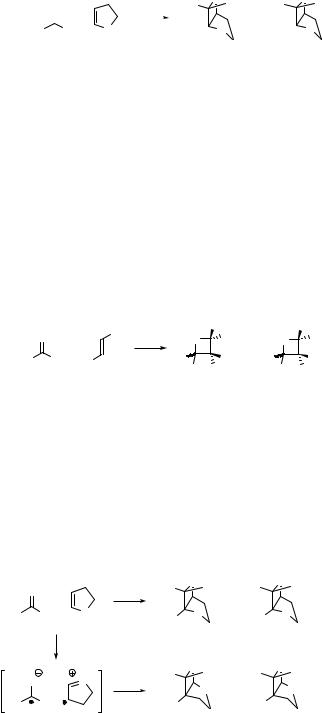
Oxygen Compounds |
305 |
medium-pressure mercury lamp (400 W) using a Pyrex filter (Figure 3.9). The reaction was monitored by TLC and was stopped after 3 h. The solvent was evaporated and the product was purified by column chromatography.
6.3.3Carbonyl Compounds: Norrish Type I Reaction
O
R 

 R
R
Recommended review articles.751,903–910
Selected theoretical and computational photochemistry references.16,187,534,535,911–916
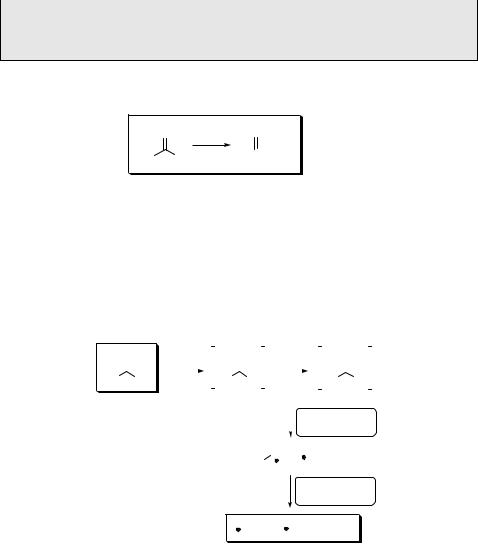
Homolytic cleavage of the a-bond (a-cleavage; Norrish type I reaction917), often
followed by decarbonylation of the acyl radical intermediate thereby formed909 (Scheme 6.109), is one of the most common reactions of excited ketones.903,905 This
reaction can be accompanied by competing processes, such as the Norrish type II reaction (Section 6.3.4) or photoreduction (Section 6.3.1).
O |
1 |
O |
* ISC |
3 |
O |
|
* |
||||||||||||
|
|
|
hν |
|
|
|
|
|
|
|
|
|
|
||||||
R1 |
|
R2 |
|
|
R1 |
|
R2 |
|
|
|
|
|
|
|
R1 |
|
|
R2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Norrish type I |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
reaction |
||||
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
R1 |
|
|
|
+ |
2 |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
decarbonylation
Scheme 6.109
There is a good correlation between the bond dissociation energies (DC CO) and the corresponding rates of a-cleavage and subsequent decarbonylation, so that the quantum yield of the reaction is directly related to the stability of the radicals formed. For example, the excitation energies of acetone (ES 373 kJ mol 1, ET 332 kJ mol 1) and acetophenone (ES 330 kJ mol 1, ET 310 kJ mol 1) are usually sufficient for exothermic release of stabilized benzyl or tert-butyl radicals (Figure 6.6, Table 6.9), whereas formation of methyl or phenyl radicals is inefficient.
In general, the n,p excited ketones undergo much faster cleavage than those with a
lowest p,p excited state, because the s-orbital of a bond being cleaved overlaps with the half-vacant n-orbital on the oxygen atom.905,911,919 The cleavage rate constant of the n,p
triplet excited benzyl phenyl ketone (PhCH2COPh) is, for example, more than three orders