

Photochemistry_of_Organic
.pdf216 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||||||||||
|
O |
|
|
|
|
3 O |
|
|
O |
||||
|
|
|
Ph |
1. hν |
T1 |
|
|
Ph |
n = 10–15 |
|
|
||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(CH2)n–5 |
2. ISC |
|
(CH2)n–5 |
|
|
(CH2)n–5 |
||||||
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|
|
n = 6–12,15 |
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
PhCH2=CH2(CH2)n–3CHO |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
(CH2)n–5 |
|
Scheme 5.16
Most interesting results were obtained by measuring the lifetimes of these biradicals in magnetic fields B ranging from 50 mT (Earth s magnetic field) to 0.1 T. The ratio of the ISC rate constants in the presence and absence of a magnetic field, kISCB =kISC0 , increased from 1 (B ¼ 50 mT) to a maximum of 1.13 and then decreased to an asymptotic value of 0.8–0.9 for B ¼ 0.1 T. The maximum was reached at 3 mT for n ¼ 12, at 12 mT for n ¼ 11 and at 60 mT for n ¼ 10. The analysis of these and other data40 proved that ISC is dominated by SOC (76% for n ¼ 12 and 88% for n ¼ 11), but that hyperfine coupling (HFC) gains importance for the longest chain lengths. It was also shown that for 1,n-dibenzyl biradicals HFC becomes dominant for n > 6, because SOC is smaller in the absence the acyl group (recall that SOC increases with the fourth power of the atomic number Z, Section 4.8).
Case Study 5.3: Mechanistic photochemistry – the photo-Favorskii reaction of p-hydroxyphenacyl compounds
p-Hydroxyphenacyl (pHP) is an effective photoremovable protecting group for anionic substrates X (see also Special Topic 6.18). The release of, for example, diethyl phosphate from compound 25 [X ¼ OPO(OEt)2, Scheme 5.17] is accompanied by a photo-Favorskii rearrangement of pHP to p-hydroxyphenylacetic acid (26), which absorbs only below 300 nm. Following fast and efficient ISC, the reaction is initiated
HO |
|
hν |
|
|
|
|
|
|
ISC |
|
–HX |
|
|
O |
|||||||
|
|
|
|
|
|
|
|
||||||||||||||
|
|
|
|
S1 |
|
|
T1 |
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
H2O |
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
X |
|
|
|
τ ≈ 4 ps |
|
|
τ ≈ 60 ps |
|
|
|
|||||||||||
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O |
|
25 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
327 τ ≈ 0.5 ns |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||
HO |
H2O |
O |
|
– CO |
O |
|
HO |
||||||||||||||
|
|
|
|
|
|
|
|
H2O |
|
OH |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
CO2H |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
26 |
|
|
|
|
|
28 τ < 0.5 ns |
|
|
|
|
29 τ ≈ 0.3 s |
30 |
|
||||||||
Scheme 5.17 Mechanism of the photocleavage of p-hydroxyphenacyl diethyl phosphate

Primary Photochemical Intermediates: Examples and Concepts |
217 |
from the lowest the triplet state T1 of 25 (lmax 390 nm). It proceeds only in aqueous solvents and the lifetime of T1 decreases in a quadratic fashion with increasing water concentration in wet acetonitrile;383 with diethyl phosphate as the leaving group X it amounts to several microseconds in degassed, dry CH3CN, but is reduced to 60 ps in wholly aqueous solution.462 Thus water plays an essential role in the elimination of the leaving group.
Time-resolved resonance Raman spectroscopy of 25 in 50% aqueous CH3CN proved that the final product 26 appears with a rate constant of 2.1 109 s 1 following pulsed excitation of 25.207 The appearance of 26 was slightly delayed with respect to the decay of T1(25), k ¼ 3.0 109 s 1, that was determined independently by optical pump– probe spectroscopy in the same solvent. The intermediate that is responsible for the delayed appearance of 26, t 0.5 ns, is attributed to the triplet biradical 327.462 It shows weak, but characteristic, absorption bands at 445 and 420 nm, similar to those of the phenoxy radical. ISC is presumably rate limiting for the decay of 327, which cyclizes to the spiro-dienone 28. The intermediate 28 is not detectable; its decay must be faster than its rate of formation under the reaction conditions. Decarbonylation of 28 to form p- quinone methide (29) competes with hydrolysis to 26 at low water concentrations. Hydrolysis of 29 then yields p-hydroxybenzyl alcohol (30) as the final product.
Detailed knowledge of this mechanism was needed to establish the release rate of the leaving group. With its fast and efficient release of diethyl phosphate, t ¼ 60 ps, F 0.4, the p-hydroxyphenacyl protecting holds promise for use in studies of fast reactions such as the primary steps in protein folding (random coil to a-helix formation) (Figure 5.18).
hν |
k |
H2O
Figure 5.18
5.4.5Carbocations and Carbanions
Recommended reviews.391, 463
Carbocations have held an important place as reactive intermediates in physical organic chemistry since the 1920s following pioneering studies of H. Meerwein, C. Ingold, S. Winstein and others. G. Olah showed that many carbocations are persistent in superacid solvents. Vinyl cations were long considered to be extremely short-lived or even nonexistent as reactive intermediates, but they are now readily accessible for study by light-induced heterolytic fission of halides, acetates and alcohols in polar solvents.464 The decay of carbocation intermediates is usually first order due to solvent addition, the rate of which increases with increasing nucleophilicity of the medium. 2,2,2-Trifluoroethanol (TFE) and 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) are especially useful for the investigation of carbocations by laser flash photolysis, because these solvents favour the efficient formation of carbocations, yet they are slow to trap them. The lifetime of carbocations is insensitive to the presence of dioxygen in solution.
218 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
|||
|
N2 |
hν |
TMB |
H |
MeO |
MeO |
|
MeO |
|
|
|
–N2 |
|
|
|
|
invisible |
|
λ max = 350 nm |
Scheme 5.18 Making photochemically generated phenyl cations visible by trapping with TMB
The highly reactive phenyl cation has long escaped detection because it does not absorb in the near-UV region; it has been identified by titration with 1,3,5-trimethylbenzene (TMB) (Scheme 5.18).465
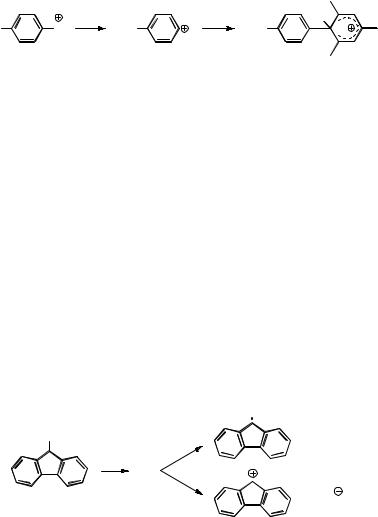
Fluorenyl cation (31), an antiaromatic carbocation, is formed by irradiation of 9- fluorenol in protic solvents (Scheme 5.19).466–469 The cation 31 exhibits a narrow
absorption band at 515 nm. The lifetime of the precursor singlet state of 9-fluorenol (lmax,abs ¼ 630 nm, lmax,em ¼ 370 nm) is strongly reduced in polar solvents, ranging from 1.7 ns in cyclohexane to <10 ps in water. The acceleration of heterolytic cleavage of the C OH bond in protic solvents must be attributed to fast proton transfer to the incipient
OH leaving group, because fission of the C OH bond occurs on a time scale that is faster (<10 ps) than that required for solvation of the ions (25 ps).468 The lifetime of the
cation 31 varies from 20 ps in water and TFE,468 to hundreds of nanoseconds in alkali
metal zeolites469 and to 30 ms in HFIP.467 |
|
|||||
The equilibrium constant for the |
hydration of 31 has been determined as |
|||||
pK |
470 |
|
equilibrium constant for the heterolytic dissociation of 9- |
|||
|
|
|||||
R ¼ 15.9. |
|
Hence the |
|
19.9 |
, which amounts to a ground-state free energy |
|
fluorenol in water is Kdiss ¼ KR/Kw ¼ 10 |
|
|
||||
difference of DrG ¼ 114 kJ mol 1.
OH |
|
+ |
OH• |
|
hν |
|
|
|
S1 |
|
|
|
polar |
+ |
OH |
|
solvents |
||
|
|
|
|
|
|
31 |
|
Scheme 5.19
5.4.6Enols
Recommended reviews.471–473
Flash photolysis has contributed much to our understanding of enol chemistry in aqueous solution. The equilibrium constant for the enolization of simple ketones, KE ¼ cenol/cketone, is usually too small permit a direct determination of enol concentrations cenol at equilibrium. For example, the enolization constant of acetone is pKE ¼ 8.3; the relative amount of 2-hydroxypropene is thus only 5 ppb in aqueous solution. Nevertheless, most reactions of carbonyl compounds proceed via enol intermediates, which, once
Primary Photochemical Intermediates: Examples and Concepts |
219 |
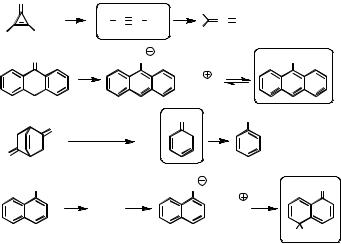
formed, are highly reactive towards electrophilic reagents. The rate law for the acidcatalysed bromination of acetone is independent of the concentration of bromine; this was observed at the beginning of the last century by Lapworth, who concluded that enolization is the rate-determining step of this reaction. Numerous kinetically unstable enols, ynols and ynamines and also keto tautomers of phenols were generated in higher than equilibrium concentrations by flash photolysis of suitable precursors and the kinetics of equilibration was monitored as a function of pH.471–473 These reactions generally obey a first-order rate law, where the observed rate constant is the sum the rate constants of ketonization and enolization, kobs ¼ kK þ kE. The technique was first applied474 to study the enol of acetophenone using the Norrish type II reaction of valerophenone (Section 6.3.4, Case Study 6.20). The same intermediate is formed by photoinduced protonation of phenylacetylene (Section 6.1.4, Scheme 6.38). Other methods that were used to generate enols include the photoreduction of ketones by hydrogen abstraction (Section 6.3.1) followed by disproportionation of the resulting ketyl radicals, the photoenolization of o-alkylphenyl ketones (Section 6.3.6) and the Wolff-rearrangement of a-diazocarbonyl compounds (Section 6.4.2, Scheme 6.171) followed by hydrolysis of the resulting ketenes via ene-1,1-diols, the enols of carboxylic acids. Some further reactions are shown in Scheme 5.20, together with examples for the generation of keto tautomers of phenols.
Rate constants for the reverse tautomerization reaction can be measured by thermal halogenation of ketones or isotope exchange reactions. Combination of the rate constants of ketonization, kE, and enolization, kK, provides accurate equilibrium constants of enolization, KE ¼ kE/kK. The acidity constants of ketones, KaK, and of the corresponding enols, KaE, are related to the enolization constant KE by a thermodynamic cycle, pKE ¼ pKaK pKaE (Scheme 5.21).
Scheme 5.21 also shows the three pathways for keto–enol tautomerization which, depending on pH, are catalysed by acid or base. The uncatalysed pathway is attributed to
|
O |
hν |
|
R |
|
|
|
|
C X , X = O, NH |
||
|
|
R C C XH |
H |
||
R |
XH |
– CO |
|
|
|
|
|
|
|
||
|
O |
hν |
O |
|
OH |
|
|
|
|
|
|
|
|
H2O |
+ H |
|
|
|
|
|
|
|
|
|
O |
hν |
O |
|
OH |
|
|
|
|
||
O |
|
– CH2=C=O |
|
|
|
|
OH |
hν |
|
O |
O |
|
|
|
|
+ H |
|
|
|
S1 |
|
|
|
|
|
H2O |
|
|
|
|
|
|
|
|
H H |
Scheme 5.20 Photochemical generation of enols, ynols, ynamines and keto tautomers of phenols

220 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||
|
|
OH |
|
|
KaK+ |
K+ |
rate determining |
|
|
|
|
|
O |
KE |
OH |
|
K |
|
E |
|
|
O |
KaE |
|
|
|
|
|
|
E– |
|
|
Scheme 5.21 Acidand base-catalysed enolization and ketonization |
||
water acting as a base, that is, E þ H2O Ð E þ H3O þ ! K þ H2O. The activation energies for unimolecular 1,3-hydrogen shifts are prohibitive, so that thermodynamically unstable enols can survive indefinitely in the absence of water and other general acid or base catalysts. The three independent reaction pathways are manifested in so-called pH–rate profiles, such as that shown for the enolization of 2,4-cyclohexadienone in Figure 5.19.475 Because 2,4-cyclohexadienone is a strong acid that rapidly protonates the solvent water, the uncatalysed reaction dominates over a large range of pH values around 7. That pathway is far less prominent in most keto–enol reactions.
The rate-determining step always corresponds to protonation or deprotonation of a carbon atom, while equilibration of oxygen acids with their conjugate bases is established rapidly. This fact can be used to determine the acidity constants of enols, ynols and ynamines by flash photolysis, KaE, either kinetically, from downward bends in the pH–rate profiles indicating a pre-equilibrium, or from the changes of the transient absorption in solutions of different pH (spectrographic titration). Such studies have provided some
remarkable benchmark |
numbers, |
such |
as the |
acidity |
constant |
of |
phenylynol |
|||||||
(pKaE |
|
2:7),476 |
phenylynamine (pKaE |
|
18:0)477 |
and its |
pentafluoro |
derivative |
||||||
E |
10:3), |
478 |
and of |
the carbon |
|
|
|
K |
¼ 2:9. |
475 |
The |
|||
(pKa |
¼ |
|
acid |
2,4-cyclohexadienone, pKa |
|
|||||||||
enolization constant of 2,4-cyclohexadienone is pKE ¼ –12.7.
Figure 5.19 pH–rate profile for the enolization of 2,4-cyclohexadienone to phenol in wholly aqueous solution at 25 C 475

Photoisomerization of Double Bonds |
221 |
5.5 Photoisomerization of Double Bonds
The E–Z photoisomerization of stilbene has been studied in considerable detail. We use the customary labels t (for trans) to designate structures near that of (E)-stilbene, c (for cis) for those near (Z)-stilbene and p (for perpendicular) for those near the transition-state geometry for E–Z isomerization in the ground state. Figure 5.20 shows schematic potential energy surfaces (PESs) for S0, S1 and T1 that are based largely on the experimental data discussed below.
Irradiation of solutions of pure c affords the cyclization product dihydrophenanthrene (DHP) with a quantum yield of about 0.10 (Scheme 6.4) in addition to t (Fc ! t ¼ 0.35).481 The Franck–Condon region reached by excitation of c to 1c* does not correspond to a minimum on the excited singlet PES. Nevertheless, solvent friction presents an effective
barrier to its rearrangement, so that the excited state 1c* persists for 0.3–2 ps depending on solvent viscosity.479,482 The initial decay of 1c* is nonexponential, exhibiting pronounced
quantum beats due to coherent wavepacket motion with a damping time of about 200 fs.483 Weak fluorescence is observed from both 1c* (Ff ¼ 8 10 5) and 1t* (Ff ¼ 7 10 5) upon excitation of c in hexane at 30 C; the fluorescence from 1t* is due to adiabatic photoisomerization 1c* ! 1t*.484 The diffuse emission spectrum of 1c* develops
vibrational structure in rigid alkane glasses at 77 K and its lifetime and quantum yield increase to 4.7 ns and Ff 0.8.485,486
Excitation of t generates 1t*, which yields c and t in approximately equal amounts, Ft ! c ¼ 0.54.487 The temperature and solvent viscosity dependence of the fluorescence quantum yields and lifetimes indicates that the reaction from 1t* proceeds over a mediumenhanced barrier in the excited singlet state; the intrinsic barrier amounts to 12 kJ mol 1.488 The fluorescence quantum yield and lifetime of t depend strongly on solvent viscosity;489 they
increase from Ff ¼ 0.035, t(1t*) ¼ 59 ps (pentane), Ff ¼ 0.040, and t(1t*) ¼ 64 ps (hexane) to Ff ¼ 0.077, t(1t*) ¼ 110 ps (n-tetradecane) at 30 C,479 and to Ff ¼ 0.95 and t(1t*) ¼ 1.6
ns in a rigid alkane glass at 77 K.488 The first absorption bands of c and t (Figure 6.1)
Figure 5.20 Potential energy surfaces479 for the E–Z isomerization of stilbene. The conical intersection480 is on the cyclization path to DHP, outside of the torsional reaction coordinate shown
222 Photochemical Reaction Mechanisms and Reaction Intermediates
correspond to the HOMO–LUMO transition, but the forbidden, doubly excited state of Ag symmetry (see end of Section 4.7) lies only 1000 cm 1 above S1(t).490
ISC to the triplet state is inefficient in media of low viscosity at ambient temperature; the triplet state must be populated by sensitization. Pulsed laser irradiation of a sensitizer in the presence of high concentrations of either c or t produces the same transient intermediate exhibiting a broad absorption band rising in intensity from 400 to 360 nm.491 The intermediate decays with a lifetime of 60 ns to give nearly equal amounts of the c and t
isomers and is attributed to the twisted triplet state 3p*. The energy of 3p* was determined by photoacoustic calorimetry to lie 195 4 kJ mol 1 above the ground state of t.251 The
reaction enthalpy for the isomerization of c to t in solution is |
Dc ! tH ¼ 19.2 |
0.4 kJ |
|||||||||||
|
1 |
and the |
|
activation |
enthalpy |
for this |
|
|
|||||
mol |
|
|
reaction in the ground state is |
||||||||||
182 |
|
8 kJ mol 1 |
492 |
|
|
|
gap nearly vanishes at perpendicular |
||||||
|
|
|
.3 |
|
Hence |
the T1–S0 |
energy |
|
1 |
p. |
|
|
|
geometries, with |
|
p* probably lying slightly below |
|
|
|
||||||||
The comparable yields of c and t formed by direct excitation of either isomer have long been taken as evidence that the E–Z photoisomerization proceeds via a common intermediate 1p*, which acts as the funnel for IC to the ground state. However, femtosecond laser studies do not appear to be consistent with such an intermediate possessing a lifetime of more than 150 fs because there is no measurable delay between the disappearance of 1c* and the appearance of ground-state product absorption.482
Theoretical calculations show that the reaction cannot be described solely by the torsional angle f.493–495 A conical intersection (Special Topic 2.5) has been located along a cyclization reaction coordinate leading to DHP.493,496 DHP opens up very efficiently on
irradiation, presumably through the same conical intersection. Contrary to an early suggestion,497 the minimum at 1p*, probably arising by admixture of the doubly excited 1 Ag wavefunction (see end of Section 4.7), is shallow and the energy gap to the ground state is fairly large at this geometry. It is therefore unlikely that 1p* represents the dominant funnel for IC to the ground state. However, in spite of the extensive work done, this issue is still under discussion.
The groundand triplet excited-state PES of the homologues 1,4-diphenylbuta-1,3- diene (DPB)498 and 1,6-diphenylhexa-1,3,5-triene (DPH)499 have also been elucidated in detail by both experiment and calculation. Stilbene and DPH represent two extreme cases because the extended conjugation in DPH lowers the triplet energies of planar conformers more than those of twisted ones. All three have rather flat triplet PESs with respect to torsional motion about the formal double bonds. The perpendicular geometry 3p* represents an energy minimum in stilbene. In contrast, the perpendicular geometry corresponds to an energy maximum in 3DPH. Decay from the 3p* state of stilbene and from roughly planar triplets in DPH accounts for the 1000-fold difference in triplet lifetimes (60 ns for stilbene, 60 ms for DPH). The roughly equal amount of (E)- and (Z)- stilbene formed by sensitization of either isomer is consistent with decay from a perpendicular triplet, whereas the partitioning from 3DPH reflects an equilibrium distribution of the planar conformers that strongly favours the all-trans triplet. Conformational equilibration is also fast in 3DPB, which decays mostly from a perpendicular minimum that lies slightly above the global all-trans minimum. Quantum chain processes of triplet–triplet energy transfer may lead to quantum yields of photoisomerization quantum yields far in excess of unity.500

Chemiluminescence and Bioluminescence |
223 |
The short lifetimes of excited diaryl polyene singlet states precludes the equilibration of conformers following excitation and, as a result, the conformational distribution in the ground state controls the product distribution. Because the absorption spectra of different conformers usually differ, this results in an excitation-wavelength dependence of the product distribution (NEER principle;501 cf. Section 6.1.1 and Case Study 6.3).
5.6 Chemiluminescence and Bioluminescence
Recommended reviews.502–509
Chemiluminescence is a fascinating phenomenon that is often displayed in demonstrations ( cold light ), seen in Nature (bioluminescence) and is widely used for analytical purposes (drug screening, DNA sequencing, capillary electrophoresis, immunoassays, forensics, etc.),506 thanks to the utmost sensitivity of light detection in a dark environment. The generation of electronically excited molecules from ground-state reagents is called chemiexcitation; it opens up the possibility of performing photochemistry without light.
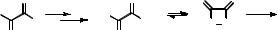
Chemiexcitation is by definition a diabatic reaction. The minimal conditions for chemiexcitation are not easily met. First, the energy released must be fairly large; the energy difference between the transition state of the thermal reaction and the final product must exceed the excitation energy of the product. The energy required to produce an excited state emitting light in the visible region of 400–700 nm amounts to 300–170 kJ mol 1. Second, an efficient pathway is needed to store the available excess energy in an electronically excited state of the product, rather than wasting it as heat, which is favoured entropically. This requires that diabatic potential energy surfaces cross along the reaction coordinate between reactant and product, such that the wavefunction of the ground-state reactant changes smoothly to that of the excited state of the product (Figure 5.21). In terms of the Landau–Zener model (Section 2.3), formation of the excited state of the product is favoured when the slope of the two zero-order PESs differs strongly at the diabatic crossing point and the electronic interaction term Vif between the two zero-order surfaces is small.
The energy requirement is an obstacle to the physical characterization of chemiluminescent compounds because the availability of a reaction of high exothermicity renders most such reagents unstable. They are usually generated in situ by mixing stable precursors with appropriate reagents. The observation of blue chemiluminescence from a solution of oxalyl chloride and hydrogen peroxide was first reported in 1963,510 now well
E |
S1 |
S1 |
S0 |
S0 |
Q |
Figure 5.21 Shape of a diabatic surface crossing favouring chemiexcitation
224 |
Photochemical Reaction Mechanisms and Reaction Intermediates |
||||
|
O H O |
2 |
O |
–H O O |
O DPA |
Cl |
2 |
|
2 |
1DPA* + 2 CO2 |
|
Cl |
HO2 |
|
O2H |
||
|
|
|
O O |
||
|
O |
|
O |
|
32 |
Scheme 5.22 The peroxyoxalate chemiluminescence reaction
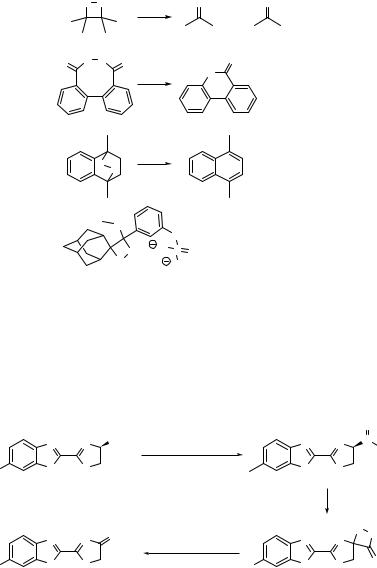
known as the peroxyoxalate chemiluminescence reaction. The authors noted: The vapors produced by the reaction have the unusual property of inducing the fluorescence of suitable indicators, e.g., a filter paper impregnated with anthracene. These observations are suggestive of a metastable excited electronic state or some other highly energetic species. The luminescence was strongly enhanced by the addition of fluorescent chromophores such as 9,10-diphenylanthracene (DPA) and the emission corresponded to the fluorescence of these substances. It took over 40 years to provide evidence that the energetic species formed in this reaction is indeed the long-suspected511 1,2-dioxetanedione (32, a dimer of carbon dioxide!) (Scheme 5.22),512 and it remains to be shown that the rise and fall of this intermediate are kinetically related to the observed rise and fall of the chemiluminescence.509, 513 High-quality ab initio calculations have shown that structure 32 represents a minimum on the PES514 but, surprisingly, a reliable calculation of the barrier to its decomposition is not available at present.
This reaction is the active principle of the cold light sticks originally marketed by American Cyanamid, now OmniGlow. The last step in Scheme 5.22 cannot be due to a simple cleavage of 32 yielding an excited CO2 molecule, the excitation energy of which is far too high. In Section 5.2, we saw that the encounter of an excited molecule with an electron donor or acceptor may lead to the formation of radical ions when the free energy of electron transfer is negative (Equation 5.1). Naturally, the reverse process may occur when the recombination of radical ions is sufficiently exergonic to form an excited state of one of the neutralized reactants. Thus, the reaction of DPA and other activators with 32 is thought to produce a radical ion pair 1(DPA. þ CO2. þ CO2), which forms 1DPA* by return electron transfer.515 Such processes were named chemically initiated electron exchange luminescence (CIEEL);516 they operate in many other chemiluminescent systems. The radical ions may also be produced by electrochemical oxidation and reduction processes, resulting in electrochemiluminescence (ECL).508 Essentially the same process is exploited in LEDs and OLEDs (Special Topic 3.2) and excimer lasers (Special Topic 3.1). Four important isolable chemiluminescent species are shown in Scheme 5.23.
The cleavage of tetramethyldioxetane (33) to two acetone molecules represents a [2 þ 2] cycloreversion reaction that classifies as ground-state forbidden by the Woodward–Hoffmann rules (Section 4.9). Hence one may expect a diabatic crossing of zero-order PESs in this reaction (Figure 4.35). Sophisticated calculations indicate that the weak O O bond breaks initially and that ISC to the triplet state occurs in the resulting biradical intermediate.517 Exothermic triplet–singlet energy transfer from the chemiexcited triplet acetone to a heavy-atom substituted chromophore (9,10-dibromoanthracene) then promotes efficient fluorescence emission.507
The majority of bioluminescent organisms live in the ocean, but there are many terrestrial forms, notably beetles, that exploit bioluminescence. The bioluminescence of fireflies
|
|
|
Problems |
|
225 |
|
O O |
∆ |
O |
O |
+ hν |
|
|
|
|
+ |
|
|
33 |
|
|
|
|
O |
O O |
O |
|
O |
|
|
|
∆ |
O |
+ |
CO2 + hν |
|
|
|
|
||
|
O |
∆ |
|
+ |
1O |
|
O |
|
|||
|
|
|
|
2 |
|
|
|
O |
O |
|
|
|
|
|
Lumi-Phos 530 |
||
|
|
O |
O P O |
||
|
|
|
|
||
|
|
O |
O |
|
|
|
|
|
|
|
|
Scheme 5.23 Isolable chemiluminescent reagents
proceeds by oxidation of D-luciferin (34) to oxyluciferin (35) that is assumed to involve a highly energetic dioxetanone intermediate (Scheme 5.24).504 The emission wavelength depends on the medium, pH and temperature. The origin of the pH dependence is far from understood.518 An enol tautomer of 35 may be involved.
|
|
|
|
|
|
O |
|
N |
N |
CO2H luciferase, ATP, Mg2+ |
N |
N |
C |
|
|
|
|
|
|
AMP |
HO |
S |
S |
|
S |
S |
|
|
|
34 |
|
|
|
|
|
|
|
|
–AMP |
O2 |
|
|
N |
N |
O |
N |
O |
O |
|
N |
|
||||
HO |
S |
S |
–CO2, –hν HO |
S |
S |
O |
|
|
35 |
|
|
|
|
Scheme 5.24 Luminescent reaction of fireflies
5.7 Problems
1.The pKa of phenalene ionizing to its anion 4 (Case Study 4.1) is about 20. The first absorption band of phenalene lies at n~0 0 ¼ 2.9 mm 1. Predict the acidity constant of phenalene in the lowest excited singlet state. [pKa ¼ 1]