

Photochemistry_of_Organic
.pdf166 |
Quantum Mechanical Models |
single-electron wavefunctions; they describe the motion of a single electron in the Þeld of stationary nuclei and the time-averaged Þeld of the other electrons in the molecule. In order to allow for the correlation of electronic movement, we must give the wavefunction more ßexibility. This can be achieved by setting up the wavefunction of an electronic state J as a linear combination of conÞgurations (Equation 4.35).
X
CJ ¼ CJK xK
K
Equation 4.35 A CI wavefunction
Once again, the wavefunction CJ is optimized by solving the secular equations, that is, by setting the secular determinant equal to zero, ||HJK Ð dJKE|| ¼ 0 (Equation 1.16, Section 1.4). The Hamiltonian operator that includes the electronic interaction terms 1/rij is used to calculate the matrix elements HJK. The index J identiÞes the wavefunctions associated with each eigenvalue EJ, which are now called state wavefunctions and represented by the capital symbol CJ. The index K of xK marks the conÞgurations formerly identiÞed by the symbol i ! j indicating the orbitals involved in singly excited conÞgurations. The procedure is called configuration interaction (CI or SCI, if only singly excited conÞgurations are included for optimization). Of course, multiply excited conÞgurations may be included in setting up the trial CI wavefunction (DCI for doubly excited conÞgurations, etc.). A disadvantage of CI wavefunctions is that they can no longer be interpreted in a transparent way unless individual states and transitions are well described by a ÔleadingÕ conÞguration, which has a large coefÞcient CJK in the CI wavefunction.
The additional freedom given to the CI wavefunction now allows for correlation and, Ôin principleÕ (i.e. if an inÞnitely large basis set of AOs and an inÞnitely large set of excited conÞgurations are used in ab initio calculations), this method provides exact solutions of
the Schr€odinger Equation 1.5. The approximations mentioned above are those included in the method developed by Pariser, Parr and Pople (PPP SCF CI method).294,295,296 Such
calculations for large organic molecules are completed in seconds on a desktop computer and are still useful for predicting and interpreting the absorption spectra of planar aromatic molecules and radicals or radical ions.
We have been able to deduce a number of useful results without considering electron correlation or even by disregarding electronic repulsion altogether. Indeed, singledeterminant SCF wavefunctions are usually similar to conÞgurations built from simple HMOs. Some of the serious deÞciencies of the HMO procedure, such as the degeneracy of singlet and triplet conÞgurations with the same occupation numbers, can be removed by qualitative consideration of the relevant K integral determined from HMOs (Equation 4.34). It turns out, however, that excited state conÞgurations calculated by the HMO or PPP SCF procedure frequently occur in degenerate pairs, not only due to molecular symmetry, but often accidentally as a consequence of the Coulson–Rushbrooke pairing theorems. In order to obtain an adequate representation of such excited states, Þrst-order conÞguration interaction between degenerate conÞgurations must be considered.
Consider the absorption spectrum of naphthalene (Figure 4.16) as an example. To predict the near-UVabsorption bands, we need to consider only the four inner HMOs c2 to c 2, which are shown on the left-hand side of Figure 4.26. On the right, the four lowest singly excited conÞgurations are shown and their energies relative to the ground state

The Need for Improvement; SCF, CI and DFT Calculations |
167 |
Figure 4.26 Inner HMOs (left) and energies of lowest configurations (right) of naphthalene. The size of the coefficients is |cjm| ¼ 0.408 in c2 and c 2 and |cjm| ¼ 0.425 or 0.263 in c1 and c 1
|
0 |
ð1 |
i ! j Þ |
ð |
1Þ ¼ |
|
|
|
1x |
|
; E 1x |
|
E |
1x0 |
«j |
|
«i are given in units of b, which is a negative quantity. Note |
that Eð x1 ! 2Þ ¼ Eð x 1 ! 2Þ due to the pairing of orbital energies (Equation 4.23). If we calculate the energies of the excited conÞguration by an SCF method (Equation 4.33), they will be shifted, but the qualitative picture remains the same. In particular, the degeneracy Eð1x1 ! 2Þ ¼ Eð1x 1 ! 2Þ will not be removed. It has been shown that the pairing theorems remain valid for PPP SCF calculations.
Because an SCF calculation does not allow for correlation, the wavefunctions of the
conÞgurations are not eigenfunctions of the Hamiltonian operator ^. We set up a CI
H
wavefunction (Equation 4.35) considering only the two degenerate wavefunctions, C ¼ C1x1 ! Ð2 þ C2x2 ! 1, and we have to solve the 2 2 secular problem (Equation 4.36).
|
Eðx1 ! 2Þ E |
E |
x2 |
H12 |
E |
|
¼ |
0 |
|
|
H21 |
1 |
Þ |
|
|
||||
|
|
ð |
|
! |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equation 4.36 First-order CI calculation
The off-diagonal elements H12 ¼ H21 calculated by the PPP SCF method are substantial, H12 ¼ 0.8 eV. The resulting split between the two formerly degenerate conÞgurations is sufÞcient to move the lower CI state below the energy of the conÞguration x1 ! 1. The result of a full PPP SCF SCI calculation (Figure 4.27) is essentially the same.
Figure 4.27 State diagram for naphthalene resulting from a PPP SCF SCI calculation
168 |
Quantum Mechanical Models |
The state symbols 1Lb, 1La, 1Bb and 1Ba shown in Figure 4.27 were introduced in 1949 by Platt.297,298 He assumed a free-electron model, similar to the electron-in-a-box, in
which the p-electrons of a cyclic system are conÞned to a one-dimensional loop of constant potential (a circular wire). The eigenvalues of a single electron in a perimeter of length l are given by Equation 4.37.
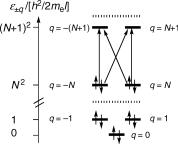
«q ¼ q2h2=ð2mel2Þ; q ¼ 0; 1; 2; . . .
Equation 4.37 Eigenvalues of an electron in Platt s perimeter potential
The orbital energies «q are all positive because the model does not contain an attractive potential. They are twofold degenerate except for the lowest one with q ¼ 0. In the closedshell conÞguration x0 of a perimeter with (4N þ 2) electrons, all orbitals up to «N and «ÐN are doubly occupied (Figure 4.28). The quantum number q represents the orbital angular momentum of an electron in orbital «q and the levels with |q| > 0 accommodate electrons moving either clockwise or counter-clockwise around the loop. Platt s original model has
been recast in terms of LCAO MO CI theory, reÞned and extended to cover a wide range of electronic spectral data.15,299 Thus, the Platt symbolsj are still in use, although the original
model is now only of historical interest.
Figure 4.28 Closed-shell ground configuration of a perimeter with 4N electrons
We return to Figure 4.27. The transition moments to the singly excited conÞgurations are readily calculated using Equation 4.20 and the HMO coefÞcients given in Figure 4.26. All four transitions are allowed and in-plane polarized (x long axis, y short axis), Mel,y(x1 ! 1) ¼ 82 e pm, Mel,x(x2 ! 1) ¼ Mel,x(x1 ! 2) ¼ 153 e pm and
Mel,y(x2 ! 2) ¼ 70 e pm. First-order mixing of the conÞgurations x2 ! 1 and x1 ! 2 gives the two states C ¼ 2 1/2(x2 ! 1 x1 ! 2). The transition moments of the two
conÞgurations cancel in the lower state and add up in the upper one. Using Equation 2.19 and the calculated transition energies shown in Figure 4.27, which are close to the experimental ones, we predict the following oscillator strengths for the four transitions:
f ð1LbÞ ¼ 0; f ð1LaÞ ¼ 0:25, f ð1BbÞ ¼ 1:3; |
f ð1BaÞ ¼ 0:17. |
|
|
|
||||||||||||||
|
|
|
1D |
q |
|
1 |
(allowed) |
for |
excitation to the conÞgurations |
1x |
N þ 1and 1x |
N 1; |
||||||
j The |
|
|
|
|||||||||||||||
|
orbital angular momentum change is |
|
¼N |
|
1 |
|
1 |
|
N |
|
1 |
|
|
N |
N |
|||
Dq ¼ 2N þ 1 (forbidden) for the transition to |
x N |
|
þ |
|
and |
|
xN |
|
|
|
. States composed of the Þrst two were given the label B |
|||||||
(starting from A for Dq ¼ 0) and L for the last two. The indices a and b refer to the polarization of the absorption bands. In molecules that contain a plane of symmetry perpendicular to the molecular plane, the subscript a designates states with the transition moment from the ground state directed through one or two perimeter atoms and the subscript b those with the transition moment cutting only across bonds (cf. e.g. Figure 4.26).
The Need for Improvement; SCF, CI and DFT Calculations |
169 |
The predicted energies, polarizations and intensities of the Þrst transitions of naphthalene are now in good agreement with experiment.299 The 1Lb absorption is very weak due to the cancellation of transition moments, « 102 M 1 cm 1. Although the excited state belongs to the B3u irreducible representation of the D2h point group (Section 4.4) and the 1Lb transition should, therefore, be x-polarized, as is the 1Bb band, the vibronic bands of the 1Lb transition are actually of mixed polarization due to vibrational intensity borrowing from higher excited states. Such bands, which are common in alternant hydrocarbons as a consequence of the pairing theorem, are said to be parity forbidden.
The intensity of parity-forbidden transitions is very sensitive to slight perturbation of the parent compound by substitution. If one calculates the effect of inductive perturbation on the transitions x2 ! 1 or x1 ! 2 by Þrst-order perturbation theory (Section 4.4, Equation 4.13), they will be shifted in opposite directions, because the orbitals involved are not paired. Even a minor imbalance in the energy of the two conÞgurations x2 ! 1 and x1 ! 2 destroys full cancellation of the two oscillator strengths in the 1Lb transition, leading to an increased intensity. For example, the transition energies in the quinolines are similar to those in naphthalene, as expected, but the intensity of the 1Lb band is increased by over an order of magnitude. The energies are not shifted much, because the charge density at all atoms of an alternant hydrocarbon remains unity in all states, including the mixed 1Lb and 1Bb states. The uneven, but opposite, charge distributions in the conÞgurations x2 ! 1 and x1 ! 2 cancel, when they are mixed 1 : 1 by CI. In general, the energies of p,p -transitions of alternant hydrocarbons are fairly insensitive to inductive perturbation.
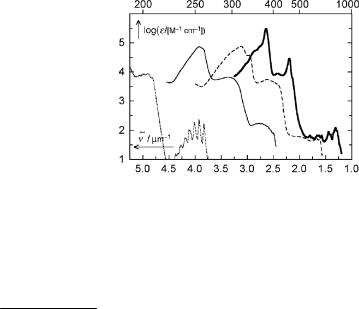
Annulenes with (4N þ 2) p-electrons, N ¼ 1, 2, . . ., are the parent representatives of Platt s perimeter model. The absorption spectra of [n]annulenes with n ¼ 4N þ 2, N ¼ 1, 2, 3 and 4, are shown in Figure 4.29.
Figure 4.29 Absorption spectra of [n]annulenes: benzene ¼ [6]annulene280 (. . .), 1,6- methano[10]annulene300 (—), [14]annulene301 (– – –) and [18]annulene302 (—)
The four lowest singly excited conÞgurations of (4N þ 2)-membered annulenes are degenerate in the HMO and Platt free electron (Figure 4.28) models. Equation 4.8 gives «LUMO «HOMO ¼ 4bsin(p/n) for the HMO excitation energies.k A PPP SCF CI
k The indices j and i for the HOMO and LUMO are n and n þ 1, respectively. Use the trigonometric equation cos a cosb ¼ 2sin g sin d, where g ¼ (a þ b)/2 and d¼(a b)/2.
170 Quantum Mechanical Models
calculation splits them into three states, which belong to the irreducible representations B2uð1LbÞ, B1uð1LaÞ and E1u(1Ba,b) of the Dnh point groups, but the positions of the three bands are still linearly related to sin(p/n).302 Transitions from the ground state (Ag) to the Þrst two are symmetry forbidden; furthermore, that to the 1Lb state is also parity forbidden. The highest energy transition to the degenerate 1Ba,b state is allowed. For the smallest member of the series, benzene, only the onset of the transition to the 1Ba,b state is seen in the deep-UV region.
More recently, the electronic spectra of the (4N þ 2) carbon cycles C10, C14, C18 and C22 were reported.303,304 The spectral pattern of C14 differs from that seen in the larger
rings, C18 and C22, indicating some sort of a structural distortion (bond length alternation). The lowest singlet state of fullerene (C60) lies at 1.57 mm 1 (639 nm) and the compound
exhibits many transitions throughout the visible and UV region, which have been assigned with the aid of calculations and symmetry arguments.305,306
Biphenylene and trisdehydro[12]annulene are representatives of conjugated hydrocarbons with a 4n-membered ring. The pattern of their absorption spectra is completely different from that of benzenoid aromatics. Their HOMO and LUMO are derived from the two NBMOs of an ideal perimeter and both the lowest excited singlet and triplet state can be described by the conÞguration xHO ! LU. The transition is symmetry forbidden in molecules of D2h symmetry or higher. Nonradiative decay usually dominates their photophysical properties. Quantum yields of ßuorescence and intersystem crossing are
low.307 The LCAO version of Platt s perimeter model has been extended to treat conjugated systems with 4N p-electrons derived from [n]annulenes.308,309
The rich absorption spectra of radical anions and cations extend well into the visible and often into the near-IR region. The radical anion and cation spectra of alternant hydrocarbons are generally very similar, nearly superimposable. This is another remarkable consequence of the pairing theorem (Section 4.6). Sketch, for example, the HMO diagram of naphthalene (Figure 4.26), add or remove an electron, respectively, and draw the arrows leading to the lowest energy excited conÞgurations in both the anion and the cation. It will become clear that for each transition of the cation there exists a transition of equal energy in the anion; for example, Eðx2 ! 1Þ of the cation is equal to Eðx 1 ! 2Þ in the anion, and so on. Hence the conÞguration diagrams for the anion and cation are identical. This is also true for the off-diagonal elements of a CI calculation, so that the absorption spectrum predicted by a PPP SCF CI calculation for the anion of an alternant hydrocarbon is identical with that of the corresponding cation. An extensive collection of radical ion spectra is available.310
Many linear polyenes exhibit anomalous ßuorescence behaviour in the sense that the ßuorescence rate constant kf calculated from the absorption spectrum (Equation 2.11) is much smaller than that determined by lifetime and quantum yield measurements (Equation 3.33). In 1972, Hudson and Kohler reported high-resolution absorption and emission spectra of all-trans-1,8-diphenylocta-1,3,5,7-tetraene at low temperature, which proved that the lowest excited singlet state S1 was not that reached by the strongly
allowed |
p,p transition (f |
|
1.5) that is predicted by MO theory and observed at |
|
410 nm. |
311 |
|
|
|
|
Rather, very weak (f 0.06), structured absorption that had been hidden |
|||
under the tail of the p,p -absorption in solution spectra was detected at slightly longer wavelengths.
The Need for Improvement; SCF, CI and DFT Calculations |
171 |
The search for a ÔforbiddenÕ state was initiated by the Þnding that the experimentally determined rate constant of ßuorescence, kf ¼ Ff/tf, was orders of magnitude less than that calculated by the StricklerÐBerg relation (Equation 2.11). This ÔforbiddenÕ state was shown to be the S1 state responsible for the long-lived ßuorescence emission. The forbidden state was immediately312 identiÞed on the basis of PPP SCF CI calculations that included doubly excited conÞgurations. The leading conÞguration of this state is 1xHO;HO ! LU;LU, that is, the conÞguration formed by twofold p,p excitation from the HOMO to the LUMO. As a conÞguration with only doubly occupied orbitals, it belongs to the same totally symmetric irreducible representation, 1Ag, as the ground state, so that the transition moment leading to the S1 state is zero.
How can a doubly excited conÞguration, xHO;HO ! LU;LU, be lower in energy than the singly excited one, xHO ! LU, when, according to simple HMO theory, EðxHO;HO ! LU;LUÞ ¼ 2EðxHO ! LUÞ ¼ 2ð«LU «HOÞ? The doubly excited conÞguration is stabilized by exchange interaction when the electronic repulsion is included in the Hamiltonian operator:
EðxHO;HO ! LU;LUÞ 2EðxHO ! LUÞ ¼ 2KHO;LU. The exchange integral KHO,LU is large in alternant hydrocarbons, whereas the energy gap «LU «HO is small in extended polyenes.
Thus, PPP SCF CI calculations place the doubly excited state slightly below the singly excited state in linear polyenes.312 The excited 1Ag state plays an important role in the photochemistry of alkenes, because it is further stabilized when the structure relaxes by twisting a formal double bond, which reduces the energy gap «LU «HO (Section 5.5).
The transition moment of the Þrst allowed p,p transition of linear polyenes is largest in the all-trans conÞguration, as predicted by HMO theory: the local charges in the transition density do not depend on the geometry, but the transition dipole is largest in the extended all-trans isomers. Isomerization of a central double bond to the Z-conÞguration is generally accompanied by the appearance of a new p,p absorption band at shorter wavelengths, the so-called cis-peak. This phenomenon has been observed in many linearly conjugated systems, including b-carotene and neolycopene,313 a-carotene314 and 1,6- diphenylhexa-1,3,5-triene.315 The appearance of the cis-peak is readily understood in terms of an HMO model that includes Þrst-order CI of the degenerate x2 ! 1 and x1 ! 2 conÞgurations (cf. the case of naphthalene, Figure 4.27). The lower of the states mixed by CI is forbidden in both isomers, but the higher lying state has a moderate transition moment that is orthogonal to the central double bond in, for example, the 3-Z isomer of hexatriene, but vanishes in the all-trans isomer.
We conclude that the HMO model, upgraded when necessary by a qualitative consideration of electronic repulsion (Equation 4.33) and of the correlation of electronic motion (by Þrst-order CI for degenerate excited conÞgurations, Equation 4.36), is able to capture the essence of electronic transitions in conjugated molecules correctly. Clearly, modern, much more sophisticated quantum chemical calculations yield more reliable quantitative predictions, but the HMO model retains its value as a language to project the output of essentially black-box calculations to a lucid formalism that provides insight, systematics and the means to generalize and extrapolate. Both the HMO and the PPP SCF CI methods are, of course, inadequate for the geometry optimization of nonplanar molecules.
Finally, we mention an entirely different quantum chemical approach, known as density functional theory (DFT). This method is designed to determine electron densities directly, rather than the electronic wavefunction itself. Knowledge of the electron density is also
172 |
Quantum Mechanical Models |
sufÞcient to deduce any observable quantity of interest. Exact DFT is an ab initio method, although most current implementations introduce some empiricism in the form of functionals such as B3LYP and M06.316 DFT methods have become very popular, because they achieve the required level of accuracy at the lowest computational cost. Excited-state energies and transition moments can be obtained using time-dependent DFT methods (TDDFT), which determine the poles of the frequency-dependent polarizability of the ground state. For these and other modern methods of computational quantum chemistry
dealing with excited states such as CASPT2,19 and a discussion of their advantages and limitations, the reader is referred to specialized texts.14,317
4.8 Spin–Orbit Coupling
Recommended reviews.318Ð320
Rate constants of intersystem crossing (ISC) are calculated by Fermi s golden rule
(Equation 2.21, Section 2.1.5). In the coupling term if ¼ < Cij^magnjCf >, Ci is the
V h
wavefunction of the initial state, for example the singlet state S1, Cf is that of the Þnal
state, in this case the triplet state T1 to which ISC occurs, and ^magn is a perturbation h
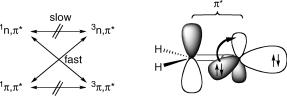
operator that accounts for magnetic interactions. SpinÐorbit coupling (SOC) is usually the dominant interaction inducing ISC, except when the radical centres are well separated in space as in extended localized biradicals (Section 5.4.4) and in radical pairs. In such systems, the magnetic interactions between electronic and nuclear spins (hyperfine coupling) become important. SOC underlies El Sayed s rules (Section 2.1.6 and Figure 4.30), which states that ISC processes involving a change in orbital type (n,p , p,p ) are generally substantially faster than those that do not. Here, we give only a brief, qualitative description of SOC. Quantitative calculations of SOC are feasible with several quantum chemistry program packages. Practical examples are discussed in Sections 5.4.1Ð5.4.4.
Figure 4.30 Left: El Sayed s rule for ISC. Right: the n,p transition of formaldehyde
The SOC operator ^SO is a sum of oneand two-electron terms. We consider only the h
more essential one-electron terms, which represent the interaction of the spin magnetic dipole moment of an electron with the magnetic dipole moment induced by its own orbital motion (Section 1.4). Approximate inclusion of the two-electron part can be done by introducing effective SOC constants z for the individual atoms, which correct for the effect of the two-electron part. The atomic SOC constants increase roughly with the fourth power of the atomic number Z. Thus, for an unpaired electron occupying an LCAO-MO, the main
Theoretical Models of Photoreactivity, Correlation Diagrams |
173 |
contributions to SOC originate from the AOs that are located on heavy nuclei and have a large angular asymmetry (p, but not s). The atomic number of protons equals 1 and nearby electrons sojourn primarily in s orbitals, so that H atoms contribute very little. The acceleration of ISC rates in the presence of heavy atoms (either in the molecule itself or in the surrounding medium, e.g. carbon tetrabromide or a xenon matrix) is called the heavy-atom effect. Rare cases of inverse heavy-atom effects are, however, also known.321 Electrons in doubly occupied orbitals, such as inner shells, can be ignored, because the contributions of the two opposed electron magnetic moments cancel. In a biradical that is well described as having two singly occupied MOs, SOC is due to the electrons occupying
these two orbitals. |
^ |
||
The SOC matrix element |
|||
< S1jhSOjT1> represents a vector consisting of three terms, |
|||
^ |
^ |
^ |
|
< S1jhSOjT1x>, |
< S1jhSOjT1y> and < S1jhSOjT1z>, because the symbol T1 comprises three |
||
states that are very close in energy and a reacting molecule can be in any one of them. The terms inducing ISC to the individual triplet sublevels generally differ from each other, so that the triplet sublevels formed by ISC from S1 will initially be unevenly populated. Rates of ISC from T1 to the ground state S0 will also differ for each sublevel. However, equilibration between the three sublevels of T1 is established and maintained within nanoseconds at room temperature and the decay of longer-lived triplet states obeys a single
exponential rate law. For equilibrated sublevels, the length of the S0ÐT spinÐorbit coupling |
|||||||
^ |
2 |
^ |
2 |
^ |
2 |
=2 |
|
vector, ½ < S0jhSOjT1x> þ < S0jhSOjT1y> þ < S0jhSOjT1z> & |
1 |
, determines the overall rate |
|||||
|
|||||||
of ISC.
The SOC operator contains the product of a term that acts on the spin part of a wavefunction, converting a triplet function to a singlet function and a term that acts on the space part of the wavefunction, changing one electronic conÞguration to another. Each
component of the SOC vector, < S0j^ SOjTlu>, ¼ , or , is a sum over all atoms and h u x y z
each atom contributes a sum over pairs of all basis set orbitals in the molecule. The terms in the sum are multiplied by numerical coefÞcients obtained from the CI expansion and from the coefÞcients of the valence (primarily p-) orbitals in the two singly occupied MOs of T1. The dominant contributors to the sum are those in which both basis set orbitals are located on the same atom (Ôone-centre termsÕ). Each term reßects the degree to which a 90 rotation around the axis u through the atom converts one member of the orbital pair into the other. If the space part of the wavefunctions differs only by the occupation number
c c < c j^ jc > of two MOs i and j, we need to consider only the three matrix elements i hSO j
(Figure 4.30).
4.9 Theoretical Models of Photoreactivity, Correlation Diagrams
When trying to predict the outcome of a photoreaction, one should certainly not let oneself be guided by the rules of ground-state reactivity. If anything, expect the opposite to happen! This sweeping statement may be justiÞed by Dewar s PMO theory (Section 4.6), which predicts an approximate mirror-image relationship between the p-electron energy of the excited state and that of the ground state. Features, such as ÔaromaticityÕ, that are taken as indicators of a stable ground state, tend to be destabilizing factors in the excited state and vice versa. Examples are provided by the heterolytic Þssion of 9-ßuorenol to form the antiaromatic ßuorenyl cation (Section 5.4.5, Scheme 5.19) and by the
174 |
Quantum Mechanical Models |
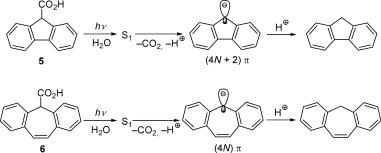
photodecarboxylation of arylacetic acids 5 and 6 (Scheme 4.2).322,323 The quantum yields and rates for photodecarboxylation are F(5) ¼ 0.042, k(5, S1) ¼ 9 106 s 1 and F(6) ¼ 0.60, k(6, S1) ¼ 6 109 s 1. The opposite would have been expected for thermal decarboxylation of 5 and 6 to form ÔaromaticÕ and ÔantiaromaticÕ anions, respectively.
Scheme 4.2 Photodecarboxylation322,323
In Section 2.1, we developed rules of thumb to predict rate constants of photophysical processes for a given molecule (Table 2.1). These unavoidable energy-wasting processes limit the lifetime of a singlet state to nanoseconds or less and that of a triplet state to milliseconds or 200 ns in aerated solutions. For a photoreaction to compete efÞciently, Arrhenius equation indicates that barriers on the excited state PES exceeding Ea ¼ 30 kJ mol 1 for S1 and Ea ¼ 60 kJ mol 1 for T1 will be prohibitive.
Once promoted to an excited singlet state by vertical excitation (FranckÐCondon principle), a molecule is likely to follow the path of steepest descent to a local minimum and relax rapidly to the lowest singlet state by internal conversion. Intersystem crossing to the triplet manifold is an option, especially for molecules with low-lying n,p states or heavy atoms. Now we are looking for guidelines to predict which reaction pathways might compete with the photophysical processes that eventually restore the starting material.
Conical intersections and avoided crossings of excitedand ground-state surfaces correspond to minima on the singlet excited PES and internal conversion to the ground state will be especially fast in such regions. These locations in the space of molecular geometries represent funnels that siphon the molecule to the ground state. The shape of the ground-state PES will then govern the subsequent reactions and may lead the molecule to a new structure, a photoproduct. Photoproducts often have a higher heat of formation than the starting material, because funnels on the excited state PES are by deÞnition at an energy maximum of the ground state. Our task is then to predict at which geometries such funnels are located and whether they are accessible from the molecular structure that is initially reached by the absorption of a photon. No funnels are expected on the lowest triplet PES, because there are no lower triplet states and diabatic ISC at crossings with a lower singlet state has a low probability (Equation 2.53). Thus, triplet states are likely to
decay more slowly by ISC from a local minimum on the triplet PES.
Modern quantum mechanics can provide accurate excited-state PESs,14,16,17 but such calculations are demanding and in order to obtain useful answers one must ask the right questions. The chemist s intuition or knowledge from related reactions is instrumental in suggesting conceivable photoproducts of the initial reactant. Qualitative guidelines to
Theoretical Models of Photoreactivity, Correlation Diagrams |
175 |
predict the initial slope on an excited PES along a chosen reaction coordinate can be obtained by Þrst-order perturbation theory using excited-state charge densities for heterolytic reactions and bond orders for homolytic reactions and rotations around formal double bonds. Thus, the changes in charge density and bond order upon excitation are most relevant.324
Early examples for the use of excited-state charge distributions to predict regioselec-
tivity are the nucleophilic aromatic substitution reactions of nitromethoxybenzenes (Case Study 6.15)325,326 and the photosolvolysis reactions of donor-substituted benzyl
derivatives (Scheme 6.147).327 In the ground state, the charge distribution of donorsubstituted benzene derivatives corresponds to that of the isoelectronic benzyl anion, which is given by the square of the NBMO [ortho- and para-activation, Figure 4.31; the saturated side-chain with a leaving group (e.g. CH2OAc) that may be attached at the ortho-, meta- or para-position is not shown]. HOMOÐLUMO excitation accumulates excess charge predominantly in the meta- and, to a lesser extent, in the ortho-position, which favours heterolytic cleavage of the leaving group.
Figure 4.31 Reorganization of the charge distribution in benzyl anion upon HOMO ! LUMO excitation. The charge distribution in the ground state is given by the square of the HOMO and that of the excited state by the square of the LUMO
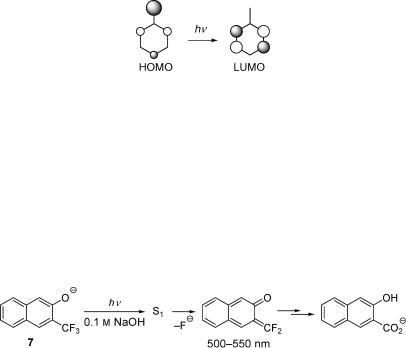
In a related reaction, the photohydrolysis of trißuoromethylnaphthols, the calculated
charge distribution in the excited singlet state was found to correlate remarkably well with the excited-state heterolysis rate constants of eight isomeric compounds.328,329 The most
reactive of these were the 1,8-, 1,5- and 2,3-substituted derivatives. The primary photoproduct formed from 3-trißuoromethyl-2-naphthol (7, Scheme 4.3), a naphthoquinone methide, was observed by ßash photolysis. Depending on the solvent polarity and on the leaving group, homolytic cleavage may, however, compete and reactions that proceed via the triplet state do not necessarily follow the same pattern.330
Scheme 4.3 Photohydrolysis of 3-trifluoromethyl-2-naphthol (7) in aqueous base
Various approximations allow one to draw diabatic surfaces that connect the electronic states of the reactant with those of conceivable photoproducts and symmetry considerations (Section 4.4) are extremely helpful in this respect. We Þrst consider state symmetry correlation diagrams.331 If the molecules of interest are symmetric (perhaps ignoring some substituents), we consider a reaction coordinate leading from reactant to