

Photochemistry_of_Organic
.pdfEnergy Transfer, Quenching and Sensitization |
55 |
singletÐtriplet absorption coefÞcients «. For R0 0.5 nm, the dipole approximation is no longer adequate and other interactions come to play (see below). On the other hand, FRET
from triplet donors with high quantum yields of phosphorescence and good overlap with the singlet absorption of the acceptor can be fairly efÞcient,104,105 notwithstanding the fact
that tripletÐsinglet energy transfer (Equation 2.41) is a spin-forbidden process. The low transition dipole of the triplet emitter 3D* is compensated by its long lifetime 3t0D.
3D* þ A ! D þ 1A*
Equation 2.41 Triplet–singlet energy transfer
F€orster radii for a number of common singlet donor and acceptor pairs are given in Table 2.2.
Table 2.2 Forster€ radii determined experimentally for various D–A pairs
Donor |
Acceptor |
|
Medium |
R0/nm |
Type |
|
|
Ref. |
||
Coronene |
Rhodamine-6G |
PMMA |
2.84 |
1 * |
! A |
106 |
||||
1D* |
||||||||||
Coronene |
Acridine Yellow |
PMMA |
3.59 |
1D* |
! A |
106 |
||||
1,12-Benzperylene |
Acridine Yellow |
PMMA |
3.34 |
1D* |
! A |
106 |
||||
Pyrene |
Perylene |
|
PMMA |
3.59 |
1D* |
! A |
106 |
|||
9,10-Dichloroanthracene |
Perylene |
|
— a |
3.78 |
3D* |
! A |
106 |
|||
Phenanthrene-d |
Rhodamine B |
CA |
4.33 |
D |
! |
A |
105,106 |
|||
12 |
TMPD þ |
b |
2 |
CH3CN |
4.0 |
1 * |
2 |
A |
107 |
|
Perylene |
|
1D* |
! |
|
||||||
Coumarin 460 |
[Ru(bpy)3] þ |
CH3CN |
4.4 |
D |
! A |
108 |
||||
aCellulose acetate.
bWurster s blue radical cation.
How does energy transfer affect the decay kinetics and overall intensity of donor ßuorescence? In rigid media or highly viscous solvents, where the position of the molecules may be considered as stationary during the lifetime of 1D* (apart from, possibly, Brownian rotation), the spectral radiant intensity In~D* due to the ßuorescence of 1D*
decays non-exponentially with time t after excitation of D by a short light pulse, as predicted by F€orster67,68 (Equation 2.42).63
In~D*
Equation
|
|
|
|
|
|
|
þ |
|
|
|
|
|
t; cA |
|
D* |
t |
tD |
cA |
|
tD |
|||
|
|
In~ |
0 e |
2cA |
q |
||||||
|
|
|
|
|
|
t |
|
t |
|
||
ð |
|
Þ ¼ |
|
ð ¼ |
0 |
0 |
0 |
|
|||
|
|
Þ |
|
|
|
|
|
|
|||
2.42 Forster€ decay kinetics in solid solutions
The lifetime tD0 is that of the donor ßuorescence in the absence of A, cA is the acceptor concentration and cA0 is the so-called critical concentration of A, deÞned by Equation 2.43.
|
|
3 |
|
|
|
R0 |
1 |
|
cA0 |
¼ |
|
¼ 4:473 10 25 |
|
|
|
mol dm--3 |
|
2p3=2NAR03 |
nm |
|||||||
|
|
Equation 2.43 |
Critical concentration |
|||||
Because the decay of 1D* is accelerated by resonance energy transfer to A, the ßuorescence quantum yield of the donor is reduced and that of the acceptor A increases
56 |
Photophysics and Classification of Primary Photoreactions |
accordingly. Assuming a statistical (Gaussian) distribution of the intermolecular distances,
F€orster derived Equation 2.44 for the relative ßuorescence quantum yield of the acceptor,
max.68
FAðcAÞ |
p |
p |
xex2 81 |
2 |
x e y2 dy9 |
||
p |
|
||||||
Fmax |
|
||||||
¼ |
< |
p |
ð |
= |
|||
A |
|
|
|
|
|||
Equation 2.44 |
: |
|
|
0 |
; |
||
Concentration quenching |
|||||||
The second term in the braces is the Gaussian error function, x ¼ cA/c0A, and FA
the ßuorescence quantum yield of A when it is either excited directly or when energy
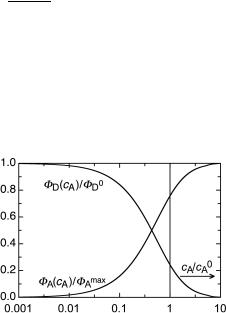
transfer from D to A following excitation of D is complete at high concentration of A. This function and its counterpart, FD(cA)/FD0 ¼ 1 Ð FA(cA)/FAmax, representing the concen-
tration quenching of 1D* by A, are plotted in Figure 2.16.
Figure 2.16 Relative quantum yields of donor (D) and acceptor (A) fluorescence in rigid solutions. The vertical bar indicates the concentration cA ¼ c0A
Thus, in rigid solutions, the critical transfer distance R0 can be determined experimentally either from the observed relative quantum yields of D or A at various concentrations of A (Equation 2.44) or from time-resolved measurement of the ßuorescence decay of D (Equation 2.42). The results are in good agreement with those calculated from the F€orster Equation 2.37.
In ßuid solutions, three regimes can be distinguished. At very high dilution, where donors and acceptors rarely approach to distances below 10 nm, radiative energy transfer dominates and the lifetime of 1D* is not affected by the presence of A. At moderate acceptor concentrations, ßuorescence quenching of 1D* by A is governed by diffusional approaches of A to 1D* . The mean molecular diffusion distance during the ßuorescence lifetime tD of the donor is given by Equation 2.45:
|
¼ p |
r |
2DtD |
Equation 2.45 Mean molecular diffusion distance
where D is the sum of the diffusion coefÞcients of donor and acceptor, which can be calculated from the StokesÐEinstein Equation 2.28.
Energy Transfer, Quenching and Sensitization |
57 |
As long as r > 3R0, the ßuorescence decay is close to exponential, the lifetime of the donor ßuorescence decreases linearly with increasing concentration of A and ßuorescence quenching obeys SternÐVolmer kinetics (Section 3.9.8, Equation 3.36). However, the bimolecular rate constants ket of energy transfer derived from the observed quenching of donor ßuorescence often exceed the rate constants of diffusion kd calculated by Equation 2.26, because resonance energy transfer does not require close contact between D and A. Finally, when r 3R0, at high concentrations and low solvent viscosity, the kinetics of donor ßuorescence become complicated, but an analysis is possible,109,110 if required.
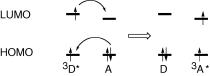
Although tripletÐtriplet energy transfer (Equation 2.46) is a spin-allowed process, total spin being conserved, it is forbidden in terms of the Coulombic (F€orster) formalism, because the oscillator strengths for singletÐtriplet absorption by A are extremely small. A quantum mechanical treatment for triplet energy transfer by electron exchange was reported by Dexter; triplet energy transfer is treated as a synchronous, dual exchange of electrons (Section 5.2) between the molecular orbitals of the donor molecule 3D* and those of the acceptor A (Figure 2.17).
3D* þ A ! D þ 3A*
Equation 2.46 Triplet energy transfer
Figure 2.17 Electron exchange mechanism for triplet energy transfer from 3D* to A. The horizontal bars represent molecular orbital energies
We need not be concerned with the quantum mechanical treatment of electron exchange. All we need to know is that triplet energy transfer by electron exchange, the Dexter mechanism, requires appreciable overlap between the molecular orbitals of D and A, so that the critical transfer distance becomes essentially equal to the sum of the van der Waals radii of D and A.
Triplet energy transfer is readily measured by nanosecond ßash spectroscopy (Section 3.7), because molecules in the lowest triplet state have strong and characteristic tripletÐtriplet absorption spectra commonly extending well into the visible region (Figure 2.18).
Triplet energy transfer is an important method used in preparative photochemistry to generate the triplet state of molecules that either do not undergo spontaneous ISC following direct excitation or that undergo unwanted reactions when excited to the singlet state. The irradiation of suitable donor molecules rather than of the target substrates directly is called triplet sensitization. The triplet energy of the donor D, ET(D) ¼ E(3D* ) Ð E(D), should exceed that of the acceptor, DET ¼ ET(A) Ð ET(D) 0 for efÞcient energy transfer. Moreover, exclusive excitation of the donor must be possible. This is best achieved if the donor absorbs at longer wavelengths than the acceptor (Figure 2.19).

58 |
Photophysics and Classification of Primary Photoreactions |
Figure 2.18 Energy transfer from triplet thioxanthone (lmax 650 nm, ET ¼ 265 kJ mol 1) to naphthalene (1 10 3 M, triplet–triplet absorption of triplet naphthalene: lmax 415 nm, ET ¼ 254 kJ mol 1) observed at various delay times with respect to a nanosecond excitation pulse at 351 nm that is absorbed only by thioxanthone
Benzophenone (ET ¼ 288 kJ mol 1) and the xanthones (ET ¼ 265 kJ mol 1 for thioxanthone, Figure 2.18) are often used as triplet sensitizers, because they absorb at relatively long wavelengths, undergo efÞcient ISC (FISC 1) and have a small singletÐtriplet energy gap. Alternatively, the donor can be used in large excess, for example acetone as a solvent, to ensure that it absorbs most of the light.
If the triplet energy of the acceptor is comparable to or higher than that of the donor, DET ¼ ET(A) Ð ET(D) 0, then the efÞciency for separation of a thermally equilibrated encounter complex with the excitation energy residing on the acceptor A is given by Boltzmann s distribution law, nA /nD ¼ exp( DET/RT), so that the rate constant ket ¼ kdnA /(nA þ nD ) is expected to fall off exponentially as energy transfer becomes strongly endothermic (DET/RT 1) (Equation 2.47). Such behaviour was observed in pioneering studies for triplet energy transfer from biacetyl (ET ¼ 236 kJ mol 1) to a graded
Figure 2.19 Triplet sensitization

Energy Transfer, Quenching and Sensitization |
59 |
series of acceptors with higher and lower triplet energies.111 |
|
||
ket ¼ |
kd |
kde DET=RT for DET |
0 |
1 þ eDET=RT |
|||
Equation 2.47
Hammond, Saltiel and co-workers subsequently observed that the rate constant of energy transfer becomes slower than that predicted by Equation 2.47 when the structure of the relaxed excited triplet state of the acceptor differs substantially from that in ground state, as is the case for stilbene.112 These Þndings initiated extensive investigations on the twisted triplet state geometry of stilbene and related compounds (Section 5.5). The expression nonvertical energy transfer (NVET) was coined to describe this situation.
A classical treatment of NVET that was gleaned from electron transfer theory (Section 5.2) was given by Balzani (Equation 2.48).113
ket ¼ |
|
|
|
kd |
|
1 |
þ |
eDG=RT |
k 0d |
eDGz=RT |
|
|
|
|
þ ket |
||
Equation 2.48
The free energy difference DG between the donor and the acceptor triplet excitation free energies is usually replaced by the spectroscopic energy difference DET ¼ ET(A) Ð ET(D) that is determined from the 0Ð0 bands of the phosphorescence spectra, ignoring the difference in entropy changes that may be associated with excitation of A and D to the triplet state (see Section 3.11). The free energy of activation DGz appearing in the denominator represents the excess free energy required for nonvertical energy transfer from D to A. It is treated as a Þtting parameter. A value of DGz ¼ 1300 cm 1 reduces the rate constant ket by about two orders of magnitude relative to kd/2 for DG ¼ 0.
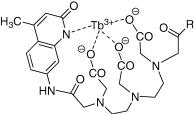
An alternative model, the Ôhot-bandÕ mechanism, found support in studies of the temperature dependence of NVET rates in stilbene sensitization.114,115 In this model,
thermal population of vibrational modes of the ground-state acceptor is assumed to provide access to geometries near to the relaxed excited state, as depicted in Figure 2.20. It is supported by the fact that energy differences between triplet donors are reßected almost entirely in the activation entropy rather than in the activation enthalpy, as would be expected from Balzani s treatment.
Figure 2.20 The energy of the donor is insufficient for vertical energy transfer to the acceptor (dashed arrow), but sufficient for thermally activated NVET. The squares of vibrational wavefunctions are shown
60 |
Photophysics and Classification of Primary Photoreactions |
Other examples of NVET include the triplet energy acceptors 1,3,5,7-cyclooctate- traene,116 which has a puckered structure in the ground state but relaxes to a planar structure in the triplet state, and a cyclobutadiene derivative,117 which changes from a rectangular to a square structure.
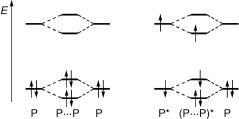
Lanthanide ions exhibit long-lived, strong luminescence in the visible region. Their fÐf absorption bands are, however, very weak and FRET to lanthanide ions is thus inefÞcient. EfÞcient FRET and even two-photon absorption118 can be achieved by complexation with appropriate chromophores as an antenna. Lanthanide ion complexes (Figure 2.21) are used
Figure 2.21 A terbium ion complex used as a probe in biophysical studies
for imaging ion channels in living cells, to study proteinÐprotein interactions or DNAÐprotein complexes in cells and for high-throughput screening assays to measure peptide dimerization associated with DNA transcription factors and ligandÐreceptor interactions.119
2.2.3Excimers and Exciplexes
The ßuorescence spectrum of dilute pyrene (<10 4 m) in degassed cyclohexane exhibits vibronic structure and has a maximum at 395 nm. As the concentration of pyrene is increased, the ßuorescence quantum yield of pyrene decreases and a broad, structureless emission band with a maximum at about 480 nm gains in intensity (Figure 2.22, left).
This phenomenon was Þrst identiÞed and explained by F€orster.120 The structureless emission is attributed to an excited pyrene dimer (1P P) that is formed by collisional association of singlet excited pyrene 1P with a pyrene molecule P in the ground state. It was subsequently found that many aromatic molecules exhibit similar behaviour. The expression excimer (excited dimer) was proposed by Stevens to distinguish such species from the excited state of a ground-state complex. Excimer formation is prominent at relatively low concentrations of pyrene (Figure 2.22, left), because of its unusually long ßuorescence lifetime, 1t ¼ 650 ns, which allows for diffusional encounters of 1P with P even at low concentration.
The diagram in Figure 2.22 (right) shows that the approach of two pyrenes is repulsive if both molecules are in the ground state, but slightly attractive when one molecule is in the excited singlet state. The vibrational states of pyrene refer to the internal coordinates of the molecule that are orthogonal to the intermolecular distance rP. . .P. They give rise to the structured emission of single pyrene molecules. The excited dimers are in a shallow potential energy minimum that has no counterpart on the ground-state surface.

Energy Transfer, Quenching and Sensitization |
61 |
Figure 2.22 Left: fluorescence spectra of pyrene in cyclohexane. Intensities are normalized to a common value of Ff. A, 10 2 M; B, 7.75 10 3 M; C, 5.5 10 3 M; D, 3.25 10 3 M; E, 10 3 M; G, 10 4 M. Reproduced by permission from ref. 109. Copyright 1970, John Wiley & Sons, Ltd. Right: potential energy surfaces for excimer formation; rP. . .P represents the distance between two pyrene molecules. The vibrational levels shown for the monomers at large separation refer to degrees of freedom other than rP. . .P
Time-resolved measurements of pyrene ßuorescence show that only the structured monomer emission is observed immediately after excitation, because the pyrene molecules are not associated in the ground state. The broad emission then grows in as the excimers are formed by diffusional encounter and the equilibrium between monomers and excimers is reached.
Why are many electronically excited molecules prone to associate with one or more ground-state molecules, whereas no such association is observed in the ground state? We Þrst recall that entropy always works against the association of two molecules, because the translational and rotational degrees of freedom of one molecule are lost. From statistical thermodynamics we can easily calculate the translational entropy of pyrene in the gas phase (p ¼ 1 bar, T ¼ 298 K), Strans ¼ 174 J K 1 mol 1. It is slightly lower in solution due to the restricted mobility, but the entropy change associated with excimer formation, DexcS, is still on the order of 100 J K 1 mol 1. Hence the enthalpy of association, DexcH, needs to be substantial to ensure exergonic excimer formation (Equation 2.49). The equilibrium constant for the formation of the pyrene excimer amounts to Kass ¼ 3.3 103M 1 in isooctane at room temperature.121
DexcG ¼ DexcH TDexcS < 0 requires DexcH < 30 kJ mol 1ðT ¼ 298 KÞ
Equation 2.49 Thermochemistry of excimer formation
This condition is relaxed when two pyrene molecules are linked covalently by a
relatively short tether such that only some rotational degrees of freedom are lost upon excimer formation (Figure 2.23).122Ð124

62 |
Photophysics and Classification of Primary Photoreactions |
Figure 2.23 Excimer formation in a covalently linked bispyrene
Quantum mechanical treatments have indicated three conceptually different types of stabilizing interactions that all contribute to the stabilization of excimers. The most important of these is called exciton interaction, which can be viewed as the stabilization energy resulting from the delocalization of the excitation energy between the neighbouring molecules. The same kind of interaction is considered in the F€orster treatment of singlet energy transfer that we dealt with in the last section. We use a trial wavefunction Cexc for the excimer that consists of an equal mixture of two wavefunctions representing the excitation being localized on one or the other of the two molecules A and B, Cexc ¼ c [CACB CA CB]. If the wavefunctions CA, CA , CB and CB are eigenfunctions of the separate molecules representing the ground and lowest excited singlet state, respectively, then the energy of the excimer can be calculated using the variation theorem (Equation 1.14), which leads to the 2 2 secular determinant (Equation 2.50).
|
H11 « |
H12 |
|
¼ |
0 |
|
H21 |
H22 |
« |
|
|||
|
|
|
|
|
|
|
|
Equation |
|
|
|
|
|
|
|
|
2.50 |
|
|
|
For A ¼ B ¼ P (pyrene), the energies associated with the two basis functions are equal, H11 ¼ H22 ¼ E(P) þ E(P ), as are the interaction terms H12 ¼ H21, which in the simplest approximation are equal to the interaction between the transition moments M0 ! 1 (Equation 2.18) of the two molecules. The secular determinant expands to the quadratic equation ðH11 «Þ2 H122 ¼ 0, which yields two exciton states (Figure 2.24), the lower of which is stabilized relative to the energy of the separated molecules by the interaction energy H12.
The S0ÐS1 transition of pyrene is fairly weak, log(«/M 1 cm 1) 2 (see Section 4.7), so that only a small exciton stabilization would be predicted by the simple treatment outlined above. In this case, higher excited states and also charge transfer states of the type
Figure 2.24 Exciton interaction
Energy Transfer, Quenching and Sensitization |
63 |
CA þ CBÐ CA CB þ must be included in calculating H12. Contributions from charge transfer states will be much more important in encounter complexes of an excited molecule with different ground-state molecules that can act as electron donors or acceptors. Such excited-state complexes are called exciplexes and will be considered in Section 5.2. Transition moments for singletÐtriplet transitions are essentially zero, so that very little exciton interaction will result in a triplet encounter pair 3A B. Indeed, few unambiguous examples of triplet excimers are known.125 The observation of excimer phosphorescence requires either very high concentrations (with the associated danger of impurity emissions) or covalent linking of two chromophores126 to reduce the entropic handicap of excimer formation.
So far we have assumed that the overlap between the molecular orbitals of the two molecules is negligible in an excimer complex. At short distances, say rP. . .P 300 pm, orbital overlap leads to further stabilization of an excimer. As can be seen from Figure 2.25, Þrst-order perturbation of the degenerate orbitals (Section 4.3) due to
Figure 2.25 Orbital diagram for the encounter of two pyrene molecules P (left, ground state; right, excimer)
intermolecular overlap does not stabilize the encounter complex in the ground state (in fact it is destabilized, because the antibonding interaction of the doubly occupied orbitals is somewhat stronger than the bonding interaction), but leads to a net stabilization of two electrons in the excimer (P P) .
2.2.4Delayed Fluorescence
The ßuorescence emission discussed so far is produced by direct excitation of a molecule M to one of its excited singlet states that, after IC to S1 when an upper singlet state was initially populated, emits prompt ßuorescence from S1 with a lifetime on the order of nanoseconds. In addition, several processes can be envisaged that permit repopulation of S1 following ISC to T1, which can give rise to emission that has the spectral characteristics of ßuorescence, but a lifetime much longer than that of prompt ßuorescence, that is, to delayed fluorescence. These processes can be sub-classiÞed as ÔP-typeÕ and ÔE-typeÕ delayed fluorescence.32 We do not consider ÔartiÞcialÕ delayed ßuorescence due to impurities, or due to ionization followed by recombination with the formation of 1M .
The classical example of E-type delayed fluorescence is that of eosin (40,50- dibromoÐ20,70-dinitroßuorescein disodium salt) in degassed solvents. The name E-type refers to eosin, which has a high quantum yield of ISC and a small singletÐtriplet energy gap, DEST ¼ 18 kJ mol 1. Thermally activated repopulation of the S1 state by reverse ISC
64 |
Photophysics and Classification of Primary Photoreactions |
from T1 is responsible for delayed ßuorescence with the lifetime of T1. The intensity of E-type delayed ßuorescence decreases strongly at reduced temperature.32 It is not observed with aromatic hydrocarbons, which have large singletÐtriplet energy gaps (Section 4.7).
An unusual example of delayed ßuorescence exemplifying El Sayed s rules (Section 2.1.6) was recently reported for the triplet sensitizer xanthone,127 which undergoes ultrafast ISC within 1 ps. Delayed ßuorescence with a lifetime of 700 ps was observed in aqueous solution. Temperature-dependent steady-state and time-resolved ßuorescence experiments indicate that the T2(n,p ) state, which is primarily accessed by ISC from S1(p,p ), is nearly isoenergetic with the S1 state. The delayed ßuorescence is
attributed to reverse ISC from T2(n,p ), in competition with internal conversion to T1(p,p ).
P-type delayed fluorescence was Þrst observed with pyrene128 (Figure 2.9) and subsequently with many other aromatic compounds.48,49 It is due to tripletÐtriplet annihilation (Equation 2.51). According to spin statistics (Scheme 2.5), one in nine encounters of two molecules 3M will give an overall singlet pair and the energy gap law will then favour formation of one singlet excited molecule 1M , provided that the combined energy of the two triplet molecules 3M is larger than that of 1M , 2ET(M) > ES(M).
3M* þ 3M* ! 1ðM . . . MÞ ! 1M* þ M
Equation 2.51 Triplet–triplet annihilation
Because two triplets 3M are required for tripletÐtriplet annihilation, the intensity of P-type delayed ßuorescence is proportional to the square of the radiant power at moderate intensity of the light source. The decay of P-type delayed ßuorescence does not follow a simple rate law. Its decay kinetics are related to those of the triplet state, of mixed Þrst and second order.
2.2.5Dioxygen
The trials and tribulations leading to an eventual understanding of photooxidation processes (Section 6.7) represent a fascinating part of the history of science.129,130 G. N.
Lewis proposed in 1924 that paramagnetic oxygen was a biradical species. Four years later, R. S. Mulliken put forth a molecular orbital description of dioxygen and predicted the existence of two low-lying excited singlet states above its triplet ground state. He assigned the known Ôatmospheric absorption bandsÕ of oxygen at Ô1.62 voltsÕ (1.31 mm 1, 762 nm) to the upper of these ÔmetastableÕ states.
In 1931, H. Kautsky claimed that the sensitized formation of a short-lived, highly active and diffusive form of molecular oxygen was responsible for the photooxygenation of dyes.131 He supported that claim with an ingenious experiment: a yellow dye and a colourless oxidation substrate (leuco-malachite) were adsorbed separately on silica gel beads and mixed. Oxidation of the colourless gel particles to develop the colour of Malachite Green took place only when the mixture was irradiated under oxygen and did not occur, when either light or oxygen was absent.
Kautsky s Ôthree-phase testÕ indicated that the excited yellow dye generated diffusible excited O2 species that were able to reach the colourless gel particles and to oxidize the leuco dye. In spite of this convincing evidence, his hypothesis was strongly challenged and