

Photochemistry_of_Organic
.pdfPhotophysical Processes |
35 |
hx0jxv0i, the overlap between the lowest vibrational level of the ground state (y ¼ 0) and a given vibrational level y0 of the excited state.
Mel;n ! m ¼ ehCel;njSrijCel;mi
Equation 2.17
The electronic transition moment can be easily calculated on the basis of further simpliÞcations that will be introduced in Section 4.4. For symmetrical molecules, group theory provides a simple tool for predicting which, if any, of the three components (x, y or z) of the electronic transition vector Mel,n ! m are different from zero (Section 4.4). The effect of vibrational overlap will be considered in Section 2.1.9.
To obtain the total intensity of an absorption band, we have to sum the individual contributions for each vibrational level y0 (Equation 2.18).
Mn ! m ¼ ehCel;njSrijCel;mi Xhx0jxy0 i
y0
Equation 2.18 Overall transition moment of an electronic transition
If some low-lying vibrational levels y > 0 of the electronic ground state are substantially populated, we must also consider contributions from Mn,y>0 ! m,y0 (so-called hot bands). Note that transitions from y > 0 to y0 ¼ 0 are located to the red of the 0Ð0 transition (y ¼ 0 to y0 ¼ 0). Hot bands can be identiÞed on the basis of their temperature dependence given by the Boltzmann population of vibrationally excited states in thermal equilibrium.
The square of the overall transition moment Mn ! m (Equation 2.18) is proportional to the oscillator strength fnm (Equation 2.19), where the frequency vnm and the wavenumber
nm are average values for the electronic transition and D is the debye unit, 1 D 3.336
~v ¼
10 30 C m. The dimension of fnm is unity.
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
2 |
|||
fnm |
|
2 |
|
Mn m |
|
|
4:7 10 |
1 |
2 |
|
|||||||
|
|
|
|
|
2 |
3 n~nm |
|
||||||||||
|
¼ |
8p mennm |
|
! |
|
|
|
|
Mn ! m |
|
|
||||||
|
|
3he |
|
|
|
mm |
|
D |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equation 2.19 Oscillator strength
The oscillator strength was originally deÞned as unity for an electron oscillating harmonically in three dimensions, an early model of an atom. It can be determined experimentally by integration of an absorption band (Equation 2.20), where « is the molar absorption coefficient.
nm ¼ |
|
|
|
ð M |
1 cm 1 mm 1 |
|
f |
4:3 |
|
10 5 |
|
«ðn~Þ |
dn~ |
|
|
|
|
|||
Equation 2.20 Experimental determination of an oscillator strength
The width of an absorption band is typically about 0.3 mm 1. Thus for a very strong transition, « 105 M 1 cm 1, the oscillator strength f is indeed on the order of 1.
2.1.5Rate Constants of Internal Conversion; the Energy Gap Law
Radiationless transitions (IC and ISC) represent a conversion of electronic energy of an initial, excited state to vibrational energy in a lower-energy electronic state (cf. Figure 2.1). Within the BornÐOppenheimer approximation (Section 1.3), radiationless

36 |
Photophysics and Classification of Primary Photoreactions |
transitions between electronic states are impossible; the potential energy surfaces represent impenetrable walls. To remove that artefact, the correlation of electronic and nuclear motions must be included explicitly in the Hamiltonian operator. Moreover, radiationless transitions involving a multiplicity change (ISC) remain forbidden unless magnetic interactions are included. These relatively small terms are introduced by timedependent perturbation theory (Equation 2.21).
ki ! f ¼ 2hp Vif2 rf
Equation 2.21 Fermi golden rule
Equation 2.21 was introduced Dirac,33 and later dubbed golden rule No. 2 by Fermi. It is now commonly referred to as Fermi s golden rule. The rate constant for a radiationless transition between an initial and Þnal state ki ! f is thus proportional to the product of two factors, the density rf of excited vibrational levels of the Þnal state that match the energy of the initial state and the square of the vibronic coupling term between the initial and Þnal
¼ hC j^jC i ^
BO state, Vif i h f , where h is a perturbation operator that couples nuclear with electronic motion to promote IC and couples electron spin with orbital angular momentum (spinÐorbit coupling, SOC) to allow for ISC. As a result, the density of states is weighted by the square of the FranckÐCondon integrals hxijxfi (see Section 2.1.9). Once the radiationless transition has occurred, intramolecular vibrational redistribution (IVR) and dissipation of the excess energy to the solvent, vibrational relaxation, are very fast, so that radiationless processes IC and ISC are generally irreversible.
Quantitative calculations of rate constants ki ! f are demanding, but the energy gap law for ÔlargeÕ molecules is well established empirically: rate constants of IC and ISC decrease exponentially as the energy difference DE between the two electronic states increases in a series of related molecules. The physical background for this has been analysed:34Ð38 There are two opposing effects: the density of vibrational states increases strongly with increasing energy gap, yet the even stronger exponential decrease of the FranckÐCondon overlap integrals hxijxfi between the vibrational wavefunction xi of the
initial state and those of the Þnal ones xf (Figure |
2.6) dominates at large energy |
χ i |
|
i |
ρf |
χf |
|
|
f |
Figure 2.6 Left: Franck–Condon integrals hxi|xfi for IC are small for large energy gaps, because highly excited vibrational wavefunctions xf oscillate rapidly and their amplitude is small in the region of overlap with xi. Right: the density of vibrational levels in the final state increases with the energy gap
Photophysical Processes |
37 |
gaps [recall the MaxwellÐBoltzmann velocity distribution r(v) of an ideal gas, where the exponential term exp( mv2/2kT) dominates over the density-of-states term v2 at high velocities v].
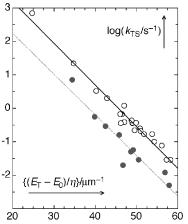
The dominant accepting modes, those vibrations that contribute most to vibronic coupling between the initial and Þnal states, are those with the highest frequency. Their FranckÐCondon integrals are comparatively large, because fewer vibrational quanta are needed to match the energy of the electronically excited state. A beautiful experimental example of both the energy gap law and the inßuence of accepting modes was provided by Siebrand39 (Figure 2.7): the rate constants for the radiationless T1 ! S0 decay of aromatic hydrocarbons, kTS, decrease exponentially with the energy gap and the rate constants for the hydrogenated compounds (n~CH 3000 cm 1) are nearly an order of magnitude larger than those of the perdeuterated compounds (n~CD 2250 cm 1). To model the observed isotope effect (see Special Topic 5.2), constant values of E0H ¼ 0.4 mm 1 and E0D ¼ 0.55 mm 1 were subtracted from the triplet energies ET (in mm 1) of the hydrogenated and deuterated compounds, respectively, and the difference was divided by the relative number of hydrogen atoms in the molecule, h ¼ NH/(NC þ NH).
Figure 2.7 Nonradiative decay rate constants of aromatic hydrocarbon triplets.39 Filled circles refer to perdeuterated compounds
The roughly exponential dependence of nonradiative transition rate constants on the energy gap DE can be turned into useful empirical rules-of-thumb to estimate rate constants of IC and ISC. As the relevant energy gaps are usually determined from absorption or emission spectra, we replace DE by Dn~, Dn~ ¼ DE/hc. Rate constants of internal conversion can then be roughly estimated from Equation 2.22.
logðkIC=s 1Þ 12 2Dn~=mm 1
Equation 2.22 Energy gap law for internal conversion
38 |
Photophysics and Classification of Primary Photoreactions |
2.1.6Rate Constants of Intersystem Crossing, El Sayed Rules
Many photoreactions of organic molecules proceed via the triplet state. Hence intersystem crossing is involved twice, Þrst upon formation of the triplet state from the excited singlet state S1 and second when the primary photoproduct, for example a triplet biradical, returns to the singlet manifold to form a stable product in the ground state. One of these relatively slow steps often determines the overall rate of the reaction. Long-lived excited triplet states are readily quenched (Section 2.2.2), allowing for a deliberate manipulation of the product distribution. ISC requires a spin ßip, which can only be induced by magnetic interactions. The magnetic coupling of electron spin to other types of motion is small. Rates of ISC are thus generally smaller than those of IC. Indeed, the electronic coupling term Vif of Equation 2.21 vanishes within the approximations used to calculate IC rate constants and spinÐorbit coupling (SOC) must be included in the Hamiltonian operator to pick up the dominant terms of these weak magnetic interactions. In biradicals with spatially well-separated radical centres, hyperÞne coupling, the interactions of electron spin with the magnetic Þeld of nuclei, may become dominant (Section 5.4.4).40
Rates of ISC are around Þve orders of magnitude slower than those of IC in organic molecules not carrying heavy atoms and also obey the energy gap law (Equation 2.23).
logðkISC=s 1Þ 7ðor 10 for n; p*,p; p*Þ 2Dn~=mm 1
Equation 2.23 Energy gap law for intersystem crossing
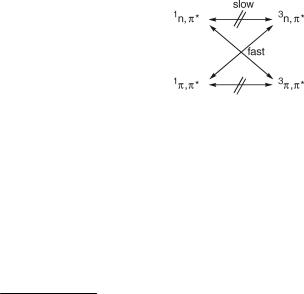
However, when ISC involves a change in orbital type,j such as in the 1n,p ! 3p,p transitions of a,b-unsaturated ketones (Figure 2.8), the reduction amounts to only about two
Figure 2.8 El Sayed rules for ISC
orders of magnitude. These qualitative rules for ISC are known as El Sayed s rules.41 Accurate calculations of the SOC term Vif appearing in the Fermi golden rule (Equation 2.21) are feasible with modern quantum mechanical software packages and the results are consistent with El Sayed s rules. Nevertheless, it is clearly desirable to have some intuitive understanding of the main factors affecting the rate constants of ISC (see Sections 4.8 and 5.4.4).
Caution is necessary when using Equation 2.23 to calculate singlet to triplet ISC rate constants, because the transition from S1 may well occur to an upper triplet state Tx rather than to T1, as long as E(Tx) E(S1). This will reduce the energy gap and may at the same
j The symmetry labels s and p are used to designate molecular orbitals (MOs) that are symmetric and antisymmetric, respectively, with respect to a plane of symmetry of the molecular structure (Section 4.4). Nonbonding s-orbitals (lone pairs) are designated with the label n.

Photophysical Processes |
39 |
time be favoured by a change in orbital type. For example, the rate constants for ISC from the S1(n,p ) states of benzophenone or 1,4-naphthoquinone are on the order of 2 1011 s 1, log(kISC/s 1) 11.3, presumably because the S1(n,p ) state couples to a nearly isoenergetic Tx state of p,p character, rather than directly to the T1(n,p ) state.42
2.1.7Quantum Yield: Definition
The quantum yield Fx(l) is equal to the amount nx of photochemical or photophysical events x that occurred, divided by the amount np of photons at the irradiation wavelength l that were absorbed by the reactant, nx/np (Equation 2.24).k Both nx and np are measured in moles or einstein (1 einstein ¼ 1 mol of photons) and the dimension of Fx is unity.
FxðlÞ ¼ nx=np
Equation 2.24 Quantum yield of a process x
An explicit deÞnition of the process x is essential to avoid confusion. For example, when x refers to a photochemical reaction A ! B, nx may be deÞned by the amount of reactant consumed (quantum yield of disappearance, FÐA ¼ nA/np),l or by the amount of product formed (quantum yield of product formation, FB ¼ nB/np). These will not be the same, FB < F A, when products other than B are also formed. In general, quantum yields lie in the range 0 Fx 1 and represent the probability that a molecule undergoes the deÞned process after absorption of a photon. However, quantum yields may exceed unity, depending on the deÞnition of process x. For a photoreaction A ! 2B, for example, the quantum yield of product formation, FB ¼ nB/np, spans the range 0 Fx 2. If the photoreaction generates radicals or other intermediates that start a chain reaction of a monomer C, R. þ C ! R C. !C RÐCÐC. !C . . ., then the quantum yield for the disappearance of C is not bounded, F C 0. When C is different from the absorbing species, F C ¼ nC/np is not strictly consistent with the deÞnition given above, because the light is not absorbed by C. By extension of Kasha s rule, quantum yields are often independent of the excitation wavelength. There are many exceptions, however. For example, when a molecule has two or more chromophores or when the sample contains a mixture of, e.g., non-equilibrating conformers, the quantum yield and the product distribution may be strongly wavelength dependent (Section 5.5).
The quantum yield of product formation FB is distinct from the chemical yield of product B. The chemical yield may approach 100% if B is the only photoproduct formed, yet FB may be low when photophysical processes dominate over product formation. Conversely, the quantum yield of reactant disappearance, F A, may be high, even though the chemical yield of product B is low, when the reaction produces mainly products other than B.
The quantitative analysis of the reaction progress is a task that chemists are well trained to do, but the determination of the amount np of photons absorbed is not. Both are needed
kWhen other absorbing species are present in the sample, then the fraction of the incident light that is absorbed by the reactant A
is AA(l,t)/A(l,t), where AA(l,t) is the partial absorbance by the reactant A (Equation 2.4) and A(l,t) is the total absorbance of the sample at the wavelength of irradiation l (Equation 2.5). Some authors deÞne np as the amount of photons absorbed by the sample (which may contain additional species absorbing at the irradiation wavelength) and consider the internal Þlter effect separately. By that deÞnition, which is not recommended, the quantum yield is no longer a property of the reactant molecule A.
lBecause compound A disappears, nA < 0, hence F A > 0.
40 |
Photophysics and Classification of Primary Photoreactions |
to deÞne the quantum yield of a photoreaction (Equation 2.24). Experimental methods to determine quantum yields and the fundamental importance of these quantities will be discussed in Section 3.9.
2.1.8Kasha and Vavilov Rules
Kasha s rule states that polyatomic molecules generally luminesce with appreciable yield only from the lowest excited state of a given multiplicity. The concept has been extended to chemical reactions of excited species, that is, polyatomic molecules react with appreciable yield only from the lowest excited state of a given multiplicity. Kasha s rule is a consequence of the energy gap law (Section 2.1.5); the energy differences between upper excited states of a given multiplicity are usually much smaller than those between the lowest singlet or triplet state and the ground state, so that the rates of IC from upper excited states far exceed the rates of ßuorescence, let alone phosphorescence. In the lowest excited states S1 and T1, radiative and nonradiative decay become competitive.
Exceptions from Kasha s rule, in particular ßuorescence from S2, are to be expected under three circumstances. First, when the energy gap between the Þrst and second excited singlet states is large, so that IC from S2 to S1 is relatively slow, then S2ÐS0 ßuorescence becomes competitive if the oscillator strength of the S0 ! S2 transition is large. Similarly,
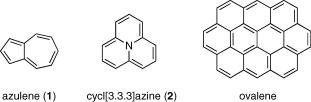
S2 S1 ßuorescence may be observed. Examples are azulene (1)43 and cycl[3.3.3]azine (2)44,45 (Case Study 4.1); for these compounds, S2 ßuorescence is much stronger than S1
ßuorescence. Second, when both the S2ÐS1 energy gap and the oscillator strength of the S0 ! S1 transition are very small but that of the S0 ! S2 transition is large, then the S1 state is likely to have a long lifetime and ßuorescence from the nearby S2 state is observed due to thermal population of the latter. This type of S2 ßuorescence, also called two-level emission, will decrease as the thermal population of S2 is reduced at reduced temperature. The classical example of two-level ßuorescence is ovalene (Scheme 2.1), which has an S2 Ð S1 energy gap of 1800 cm 1 in the gas phase and only about 500 cm 1 in toluene solution.46,47 Third, weak ßuorescence from upper excited singlet states can be detected in general with sufÞciently high sensitivity when stray light sources are carefully eliminated. This has been elegantly achieved by using tripletÐtriplet annihilation rather than direct
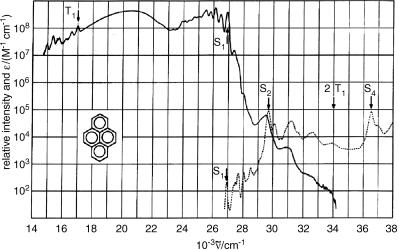
excitation to observe delayed ßuorescence (Section 2.2.4) from several aromatic hydrocarbons.48,49 The total emission spectrum of pyrene is shown in Figure 2.9. It is
well worth studying this astounding piece of work that displays ßuorescence from several upper excited singlet states in addition to phosphorescence from T1 in solution.
Scheme 2.1 Compounds exhibiting S2 fluorescence in violation of Kasha’s rule
Vavilov s rule states that the quantum yield of luminescence is generally independent of the wavelength of excitation. The quantum yield of luminescence is equal to the
Photophysical Processes |
41 |
Figure 2.9 Delayed luminescence (solid line) from a solution of 1 10 5 M pyrene in methylcyclohexane at 193 K and absorption spectrum (dotted line). Note the logarithmic scale covering eight orders of magnitude in emission intensity. Reproduced from ref. 39 with permission. Copyright 1967, American Institute of Physics
probability that the absorption of a photon by a molecule will be followed by emission of a photon from that molecule as either ßuorescence or phosphorescence. Vavilov s rule may be considered as a corollary of Kasha s rule, which implies that molecules that are excited to upper states will quantitatively relax to the lowest excited state by IC and vibrational relaxation. There are exceptions to Vavilov s rule. For example, the ßuorescence of benzene vapour suddenly drops to near zero due to some very fast radiationless decay process, the so-called channel three,50 when the photon energy is more than 3000 cm 1 above the origin of the S0 ! S1 transition.
2.1.9Franck–Condon Principle
The Franck–Condon principle assumes that an electronic transition occurs without changes in the positions of the nuclei in the molecular entity and its environment. Being much lighter than atoms, electrons move much faster. Thus the change in electronic structure (electron redistribution) associated with electronic excitation may be regarded as occurring in the Coulombic Þeld of frozen nuclei. The resulting state is called a FranckÐCondon state, an electronically excited state that is born with the structure of the ground state and the transition is called a vertical transition.
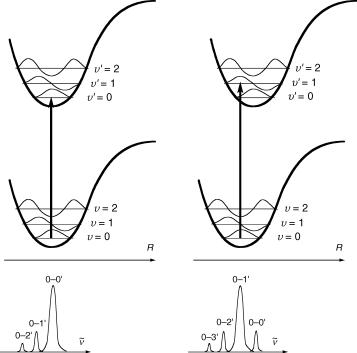
The quantum mechanical formulation of this principle was given in Section 2.1.5; the intensity of a vibronic transition is proportional to the square of the FranckÐCondon integrals between the vibrational wavefunctions of the two states that are involved in the transition. Thus, the band shape of an electronic transition depends on the displacement of the excited electronic state relative to that of the ground state. This is illustrated for one vibrational degree of freedom of a given molecule in the schematic diagram in Figure 2.10.
42 |
Photophysics and Classification of Primary Photoreactions |
Figure 2.10 Illustration of the Franck–Condon principle. The bottom diagrams illustrate the vibrational structure of the absorption bands
When the displacement of the upper electronic PES is small (left), then the overlap between the lowest vibrational levels y ¼ 0 in the ground state and y0 ¼ 0 in the excited state is largest and the 0Ð00 transition is by far the most intense. With increasing displacement, the overlap between the ground state vibrational wavefunction y ¼ 0 and the higher vibrational levels of the upper state (y0 ¼ 1 in Figure 2.10, right) becomes largest and the maximum vibronic band intensity moves to higher wavenumbers n~.
The position of the 0Ð00 band deÞnes the energy of the excited state relative to that of the ground state, E0Ð0 ¼ hn0Ð0. It can usually be located accurately in gas-phase spectra, especially in high-resolution spectra that can be obtained in low-temperature molecular beams. In solution, however, many molecules do not exhibit any vibrational Þne structure in their electronic absorption spectra, so that it is difÞcult to determine n0Ð0. Moreover, the intensity dependence illustrated in Figure 2.10 holds only for symmetry-allowed transitions (see Section 4.4). That symmetry-forbidden transitions are observable at all as weak absorptions is due to vibrational borrowing; vibronic transitions to upper (nontotally symmetric) vibrational levels become weakly allowed when the total symmetry of the vibronic transition is considered. Forbidden 0Ð0 bands are sometimes (barely) detectable in solution spectra due to symmetry perturbations induced by the solvent, but possible contributions from hot bands (Section 2.1.4) must be taken into account.
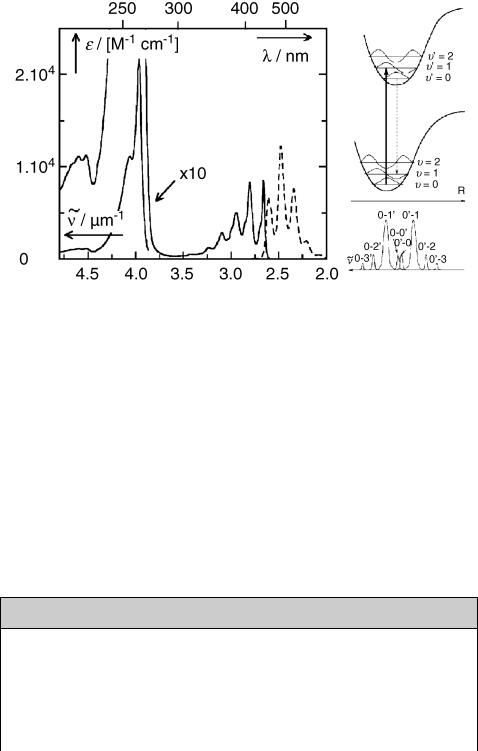
The spectral distribution of ßuorescence usually exhibits an approximate mirror-image relationship to the Þrst absorption band when the spectra are plotted on a frequency or wavenumber scale. This is expected from the FranckÐCondon principle, when the
Photophysical Processes |
43 |
Figure 2.11 Absorption and fluorescence emission of anthracene
potential energy curves of the ground and excited state are similar in shape, so that the spacing of the dominant vibronic bands is about the same in absorption and emission, as shown in Figure 2.11, right. When diffuse absorption bands do not allow an unambiguous location of the 0Ð00 band, then one usually assumes that the crossing point between absorption and emission (normalized to the same height) deÞnes the position of the 0Ð00 band and hence the excitation energy (see, e.g., Case Study 5.1).
The spectral positions of the 0Ð00 bands of absorption and ßuorescence usually do not coincide exactly. The difference is called the Stokes shift. The excited state is generally more polarizable and often more polar than the ground state. Solvent relaxation then stabilizes the excited state following FranckÐCondon excitation, so that the 0Ð00 band of absorption is usually at higher frequency than that of emission. When the excited state is much less polar than the ground state, the 0Ð00 band of ßuorescence may appear at higher frequency than that of absorption (anti-Stokes shift). Unfortunately, the term Stokes shift is also used to refer to the gap between the band maxima of absorption and emission (see the footnote to ßuorescence in Section 2.1.1), which may be much larger than the shift between the 0Ð0 bands.
Special Topic 2.2: Optical brighteners
Fluorescent whitening agents (FWAs) are important industrial products that are widely used to cover up the yellow tint of laundry, paper and road markings to produce a Ôwhiter than whiteÕ effect.51 They are also used in polymer sheets operating as concentrators of solar light. FWAs are dyes that absorb in the near-ultraviolet region and ßuoresce in the visible region, usually the blue. Fluorescent dyes absorbing and emitting at longer wavelengths are used to enhance the visibility of signposts on ski slopes or as marker inks to emphasize text. For example, a solution of ßuorescein
44 |
Photophysics and Classification of Primary Photoreactions |
appears to be green due to its strong ßuorescence; in fact, its colour is yellow when the light source is viewed through the solution. The presence of FWAs on paper and laundry becomes particularly obvious under UV illumination such as under the Ôblack lightÕ used in bars or for the analysis of thin-layer chromatographic plates. Banknotes usually do not contain optical brighteners except for security marks, so a common method for detecting forged notes is to check for ßuorescence.
Desirable properties of FWA are high absorbance in the near-UV region, but not in the visible region, that is, a strong absorption band rising sharply below 400 nm, a high ßuorescence quantum yield and therefore low quantum yields of ISC and IC and high photostability in addition to the other properties required for industrial application such as good adsorption and strong adherence to the substrates, low toxicity and rapid degradation in the environment.
The most common classes of chemicals used as FWAs are stilbenes and styrenes, triazines, benzoxazoles, coumarins, naphthalimides and covalently linked or fused combinations thereof (Scheme 2.2). DSBP, a distyrylbiphenyl derivative, is one of the most widely used FWAs in laundry detergents with an estimated worldwide production of 3000 tons per year in 1990. Its photochemical degradation by Fe(III) ions in aqueous media has been studied.52
Scheme 2.2 Typical commercial optical brighteners
A recent development is the use of quantum dots (see Special Topic 6.30) as optical brighteners in Þbres.
2.2 Energy Transfer, Quenching and Sensitization
2.2.1Diffusion-Controlled Reactions in Solution, Spin Statistics
Recommended review.53
Electronically excited states are highly reactive and so are many of their primary photoproducts. Therefore, intermolecular processes that occur on every encounter between reactant molecules in solution, also called diffusion-controlled reactions, are fairly common in photochemistry.
Encounter frequencies between solute molecules are lower than collision frequencies in the gas phase, because the translational motion is hindered by the bulk of solvent molecules. Conversely, once reactants have encountered each other in solution, they