

Photochemistry_of_Organic
.pdf256 |
Chemistry of Excited Molecules |
thick-layer chromatography or column chromatography followed by bulb-to-bulb distillation.
|
N |
hν |
|
N |
|
|
|
N |
CH3 |
Ph |
CH |
Ph |
|
|
|
|
|||
H C |
H C CH |
3 |
|
|
|
|
|||
|
3 |
3 |
|
2 |
|
|
|
|
|
|
106 |
|
|
|
|
|
107 |
|
|
Ph |
|
hν |
Ph |
Ph |
|
|
Ph |
|
|
|
|
|
|
|
|||||
|
N |
|
|
N |
H C N |
CH |
3 |
H C |
N CH |
H3C CH3 |
|
H2C CH3 |
3 |
|
3 |
3 |
|||
|
108 |
|
|
|
109 |
|
110 |
||
Scheme 6.44
6.1.5Alkenes and Alkynes: Photocycloaddition Reaction
hν
+
Recommended review articles.532,585,588,602,680–694
Selected theoretical and computational photochemistry references.16,480,534,535,695–698
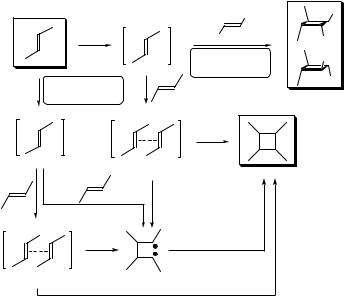
Photoinduced [2 þ 2] cycloaddition (Section 4.9) of alkenes (alkynes) to form cyclobutane (cyclobutene) derivatives is one of the best studied reactions in photochemistry.680,682 According to the Woodward–Hoffmann orbital symmetry rules,336 the cycloaddition of one singlet excited (S1) and one ground-state alkene is
allowed by a suprafacial–suprafacial concerted stereospecific pathway (Scheme 6.45).695,699,700 Rare concerted [4 þ 2] and [4 þ 4] photocycloadditions of
conjugated singlet excited dienes must occur in a suprafacial–antarafacial and suprafacial–suprafacial manner, respectively.690 Since the suprafacial–antarafacial reactant approach is geometrically difficult to achieve, [4 þ 2] reactions usually proceed stepwise (involving biradical intermediates). [2 þ 2] or [4 þ 4] photocycloadditions can occur in either a concerted or stepwise fashion.
Intersystem crossing from the excited singlet state of simple alkenes is inefficient.
Triplet state cycloadditions, therefore, are usually achievable via triplet sensitization rather than direct irradiation.701,702 Such a process often involves exciplexes, formed
between electron-poor and electron-rich alkenes,703 or 1,4-biradical intermediates (Scheme 6.45).704 Thus the tendency to achieve loose geometries in such species then favours a nonconcerted (stepwise) pathway, in which rotation about the central C C bond occurs, eventually leading to loss of reaction stereospecifity. In general, the cycloaddition
Alkenes and Alkynes |
257 |
reaction of excited alkenes can be accompanied by other processes, such as radiationless decay or E–Z isomerization (Section 6.1.1). In contrast, alkenes conjugated to a carbonyl group (enones) undergo the [2 þ 2] cycloaddition with ground-state alkenes to form cyclobutanes via the triplet state available by direct (unsensitized) irradiation (see the following Section 6.1.6).
|
|
|
|
R |
|
hν |
1 |
* |
R |
|
|
|||
|
|
|
|
concerted |
|
|
|
|
cycloaddition |
hν |
sensitized |
|
|
|
sens |
cycloaddition |
|
|
|
|
3 |
* |
1 |
* |
|
|
|
||
|
|
|
|
exciplex |
3 |
|
* |
|
|
|
|
|
|
|
|
|
exciplex |
|
1,4-biradical |
Scheme 6.45
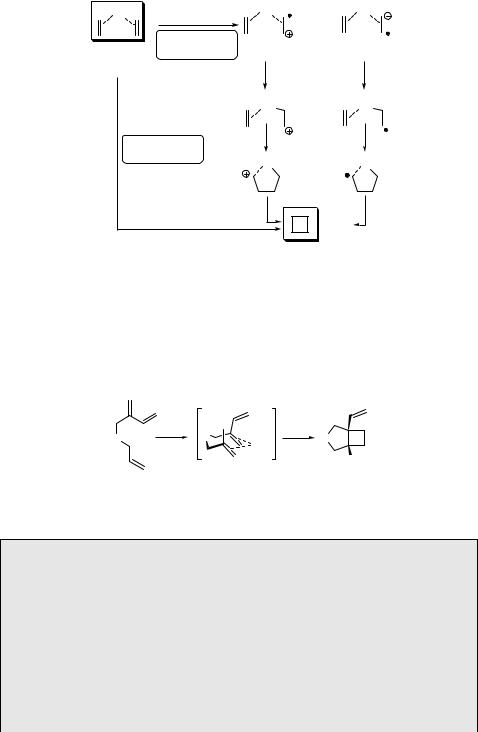
Transition metals, especially copper(I), can catalyse intermolecular or intramolecular photocycloaddition reactions of alkenes.684,688 It has been suggested that the reaction involves coordination of both the reacting alkene molecules in their ground states with a single copper(I) cation. The subsequent photoexcitation of this complex leads to cyclobutane with regeneration of the Cu(I) catalyst, probably via either metal to alkene or alkene to metal charge transfer (Scheme 6.46).705 A concerted reaction mechanism has also been proposed.706 Alkene Cu(I) complexes have been detected and isolated: whereas many aliphatic alkenes and also Cu(I) salts are almost transparent above 240 nm, intense UV absorption bands in this region are observed for their mixtures.707
There are only a limited number of intermolecular [2 þ 2] photocycloaddition reactions known to occur in solution because alkene excited singlet state lifetimes are very short (on the order of 10 ns).708 Direct irradiation of neat but-2-ene, for example, yields tetramethylcyclobutane with stereochemistry suggesting the concerted mechanism (Scheme 6.45).709 Inefficient dimerization (F ¼ 0.04), however, competes with efficient E–Z isomerization (F ¼ 0.5).
258 |
|
Chemistry of Excited Molecules |
|
||
|
Cu+ |
hν |
Cu |
Cu2+ |
|
|
|
or |
|
||
|
|
|
|
||
|
|
charge-transfer |
|
|
|
ground-state |
pathway |
|
|
||
complex |
|
|
|
||
|
|
|
Cu |
Cu+ |
|
hν |
concerted |
|
|
||
cycloaddition |
Cu |
Cu+ |
|||
|
|||||
|
|
|
|||
|
|
|
|
+ Cu+ |
|
Scheme 6.46
In contrast, copper(I)-catalysed intramolecular cycloaddition of the triene 111 affords 1-vinyl-3-oxabicyclo[3.2.0]heptane (112) in >80% chemical yield.710 Only double bonds coordinated to the catalyst participate in the photocycloaddition; the bridged arrangement of parallel constrained C¼C bonds shown is preferred over all other possible modes of coordination (Scheme 6.47).
|
hν |
O |
H |
|
|
O |
|
O |
|||
Cu+ |
Cu+ |
||||
|
|
|
|||
|
|
|
|
H |
|
|
111 |
|
|
112 |
Scheme 6.47
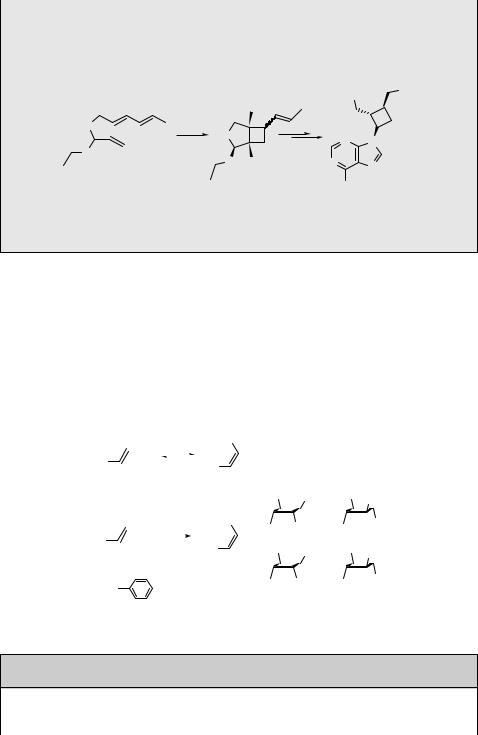
Case Study 6.10: Organic synthesis – copper(I)-catalysed photocycloaddition
The synthesis of cyclobut-A (113), a nucleoside analogue of the potent antiviral antibiotic oxetanocin, was accomplished using intramolecular [2 þ 2] cycloaddition catalysed by Cu(I) ions.711 An inseparable mixture of four (E, Z, syn, anti) isomers of 114 was obtained in 70% chemical yield by irradiation of a triene 115 (Scheme 6.48). The nucleoside connection was then established via nucleophilic substitution of an acetate group by adenine and other transformations in several steps.
Experimental details.711 A diethyl ether solution of the triene 115 (10.8 mM) and copper(I) triflate (0.94 mM) was irradiated with a medium-pressure mercury lamp
Alkenes and Alkynes |
261 |
Case Study 6.11: Hologram production – single-crystal-to-single-crystal photoreaction
A photochemical forward and thermally reversible single-crystal-to-single-crystal [2 þ 2] photodimerization reaction (such a process is termed photochromic; Special Topic 6.15) was observed on the styrylpyrylium triflate 119 (Scheme 6.51).716 The choice of the irradiation wavelength was the key parameter to a successful experiment. Intensive photolysis of a crystal using lmax close to the absorption maximum ( 420 nm) led to its breakdown; microcrystalline dimeric aggregates quickly appeared on the surface and the reaction environment turned out to be heterogeneous. However, using wavelengths within the absorption tail (lirr > 570 nm) at 20 C allowed the single crystalline character of the sample to be retained during the photodimerization. At temperatures above 100 C, the dimerized compound quantitatively reverted back to the original monomer, again retaining its crystal structure (see Special Topic 6.5). It was demonstrated that thick volume holograms can be reversibly written into such single crystals. Single-crystal-to-single-crystal reactions are feasible only when the transformations do not distort the original reactant crystal lattice too much.
|
t-Bu |
|
CF3SO3 |
t-Bu |
|
t-Bu |
|
|
|
|
|
|
O |
|
|
O hν |
|
|
|
MeO |
MeO |
|
OMe |
|
∆ |
O |
|
t-Bu |
|
|
|
|||
|
t-Bu |
|
|
|
|
CF3SO3 |
t-Bu CF SO |
|
|
|
|
|
||
|
119 |
3 |
3 |
|
|
|
|
|
|
Scheme 6.51
Experimental details.716 Grating of a hologram on a single crystal made of 119 was achieved using an He–Ne laser (632.8 nm; 50 mW) (Special Topic 3.1) for both writing and reading. Crystallographic data were obtained for monomer, dimer and mixed crystals that were isolated after different irradiation times.
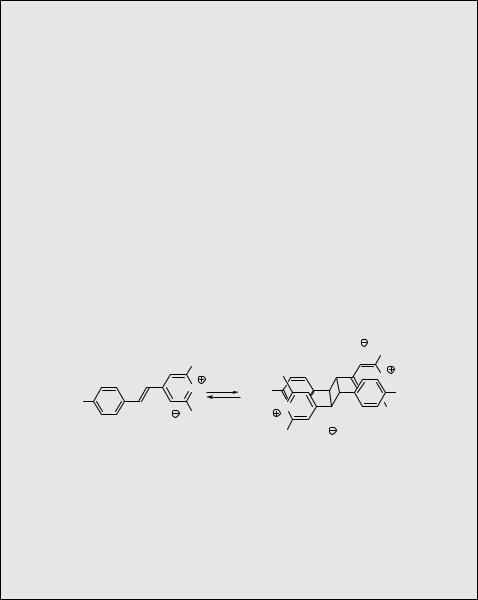
Aromatic alkynes, such as l-naphthylbuta-1,3-diyne (120) in the presence of 2,3- dimethylbutene, undergo a [2 þ 2] photocycloaddition reaction to form a cyclobutene derivative 121 in 77% chemical yield (Scheme 6.52). This particular addition occurs exclusively on the C1 C2 triple bond. Quenching, fluorescence and exciplex emission studies suggested that this reaction proceeds from both singlet and triplet excited states.717
262 |
Chemistry of Excited Molecules |
|
||
CH3 |
1 H3C |
* |
3 H3C |
* |
|
|
|
||
hν
+
120
H3C |
H3C |
121
Scheme 6.52
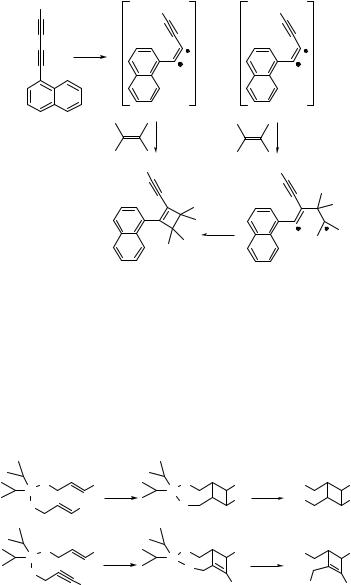
The intramolecular [2 þ 2] cycloaddition reaction in tethered alkenes or alkynes is more accessible because of favourable entropic factors. Two examples of silyl-tethered derivatives with complete regiocontrol are presented in Scheme 6.53. The compounds 122 and 123 form cyclobutane718 and cyclobutene719 derivatives, respectively, in very high chemical yields (80–90%). The tethered photoproducts are then desilylated with ammonium fluoride to obtain the corresponding diols. The photocycloaddition proceeds through the excited singlet state, as indicated by the high reaction diastereoselectivity.
Si O |
Ph |
Si O |
Ph |
HO |
Ph |
O |
hν |
O |
Ph NH4F |
HO |
Ph |
Ph |
|
|
|
||
122 |
|
|
|
|
|
Si O |
Ph hν |
Si O |
Ph NH4F |
HO |
Ph |
O |
|
O |
|
|
|
|
|
|
|
|
|
123 |
Ph |
|
Ph |
HO |
Ph |
|
|
|
|
|
Scheme 6.53
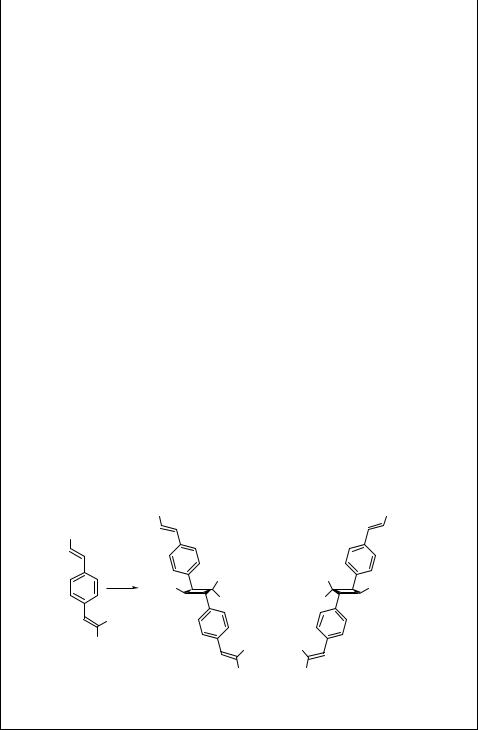
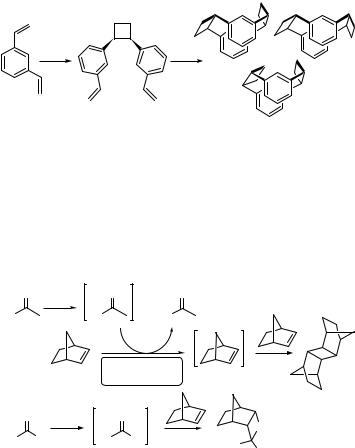
The [2 þ 2] cycloaddition reaction is an excellent method for the synthesis of cyclophanes.691 For example, irradiation of the commercially available m-divinylbenzene (124) in dry benzene yields the cyclobutane 125, which then slowly photocyclizes to isomeric cyclophanes in a low chemical yield (<10% each) (Scheme 6.54).720
|
Alkenes and Alkynes |
263 |
|
|
+ |
hν |
hν |
|
|
|
+ |
124 |
125 |
|
|
Scheme 6.54 |
|
Triplet sensitization of alkenes, such as norbornene (ET ¼ 314 kJ mol 1), having triplet energies lower than or close to that of a sensitizer, such as acetophenone (ET ¼ 10 kJ mol 1), leads to the [2 þ 2] cycloadduct 126 (Scheme 6.55).721 In contrast, oxetane 127 formation (Section 6.3.2) is observed when benzophenone (ET ¼ 288 kJ mol 1), being unable to transfer energy efficiently, is used.722
O |
3 |
O |
* |
O |
|
1. hν |
|
||||
|
|
|
|
|
|
Ph |
2. ISC |
Ph |
|
Ph |
|
|
|
|
|
3 |
* |
|
|
|
|
|
|
|
|
|
energy |
|
|
|
|
|
transfer |
|
|
O |
|
3 |
O |
* |
126 |
1. hν |
O |
||||
Ph |
Ph 2. ISC |
Ph |
Ph |
|
|
|
Ph |
||||
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
|
127 |
Scheme 6.55
Isoprene (128) undergoes [2 þ 2], [4 þ 2] and [4 þ 4] photocycloaddition reactions in the presence of a sensitizer (sens, Scheme 6.56). The product distribution is strongly dependent on the ET of the sensitizer, which is explained by the fact that the s-cis- and s-trans-conformers of 128 differ in their triplet energies and their subsequent radical (stepwise) reaction preserves their original geometries.723 A higher population of the excited s-cis-128 is then reflected in higher production of [2 þ 2] and [4 þ 4] cycloadducts.
Either direct or triplet-sensitized irradiation of cyclohexene produces a stereoisomeric mixture of [2 þ 2] dimers in nearly quantitative chemical yields (Scheme 6.57).557 Dimerization is interpreted in terms of initial photoinduced E–Z isomerization of cyclohexene (Section 6.1.1), followed by a non-stereospecific ground-state addition of (E)-cyclohexene to the Z-isomer. In contrast, copper(I)-catalysed photodimerization of cyclohexene produces only one major stereoisomer 129 (Scheme 6.57).706 According to

264 |
Chemistry of Excited Molecules |
the orbital symmetry rules, formation of this product necessitates a formal twisting of the C¼C bonds of both of the cycloalkene monomers. It has been suggested that this photodimerization involves an initial copper(I)-catalysed E–Z isomerization producing the coordination-stabilized (E)-cyclohexene 130 and supra-antara stereoelectronic control in the following addition step.
+ |
+ |
[2+2]- |
|
cycloadducts |
|||
hν |
+ |
[4+2]- |
|
+ |
cycloadducts |
||
sens |
|
||
|
|
||
128 |
|
|
|
+ |
+ |
[4+4]- |
|
cycloadducts |
|||
|
|
Scheme 6.56
+
hν
(sens)
+
hν |
Cu+ |
Cu+ |
(major) |
|
|
130 |
129 |
Scheme 6.57
Special Topic 6.6: Photochemical synthesis of cage compounds
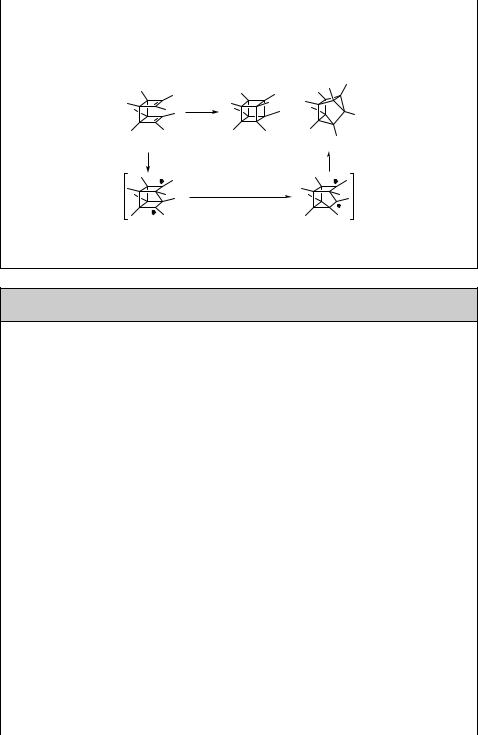
Highly strained cage compounds have long attracted considerable attention from both experimentalists and theoreticians.724 The most important structural component of cage compounds is very often a highly strained cyclobutane ring, which is easily accessible by a photochemical route.725 Cubane for its aesthetic beauty counts as one of the milestones in synthetic (photo)chemistry and structural organic chemistry.726 A
Alkenes and Alkynes |
265 |
small amount of octamethylcubane (131) was, for instance, obtained by direct irradiation of syn-octamethyltricyclo[4.2.0.02,5]octa-3,7-diene (132) via cycloaddition in n-pentane along with the major product octamethylcuneane (133), formed possibly through a stepwise radical head-to-tail bond closure (Scheme 6.58).727
hν
+
132 |
131 |
133 |
Scheme 6.58
Special Topic 6.7: Photochemistry of DNA
The UVB region of the solar spectrum (290–320 nm) is effectively filtered out by the ozone layer in the stratosphere; nevertheless, residual radiation that penetrates to Earth s surface still remains harmful to all organisms, increasingly so as the ozone layer is partially depleted. Nucleic and amino acids present in the skin are considered important UVB chromophores due to their absorbance below 320 nm. DNA bases have short S1 excited-state lifetimes (1 ps or less), which largely protects them against
greater photochemical damage, which would occur particularly when they undergo intersystem crossing to the reactive triplet states.728,729 Instead, they rapidly relax to the
ground state by nonradiative transition.730,731 Prebiotic chemistry most probably took place in an oxygen-free atmosphere and, hence, ozone-free stratosphere. Therefore, under conditions of intense short-wavelength irradiation, the photochemical stability of key building blocks probably played an important role in early chemical evolution.732 Photodimerization of adjacent pyrimidine bases (cytosine and pyrimidine) is
undoubtedly the most common photoreaction resulting from UV irradiation of DNA.728,729,733,734 This reaction has been correlated with cell survival, mutagenesis
and the development of skin cancer. The major reaction is a [2 þ 2] photocycloaddition to form a cyclobutane pyrimidine dimer (e.g. the cytosine dimer 134) via the excited triplet state735 (Scheme 6.59) or the Paterno`–B€uchi reaction (Section 6.3.2) to give the dimer 135 (Scheme 6.60). The formation of dimers causes a structural distortion of a double-helical DNA and a considerable destabilization of the duplex around the lesion, which has to be repaired to avoid cell death or mutagenesis.728 A number of different enzymatic pathways of repair have evolved. In a process called photoreactivation, the enzyme photolyase binds, for example, to the dimer and catalyses the cleavage of the corresponding bonds upon exposure to near-UV and visible light (300–600 nm) to restore the original DNA molecule.