376 |
Chemistry of Excited Molecules |
the molecule, the position of the methyl group determines its most stable shape (having the methyl substituent in a pseudo-axial orientation), which subsequently undergoes photoisomerization. As a result, it determines whether clockwise or anticlockwise rotary motion occurs.
Scheme 6.206
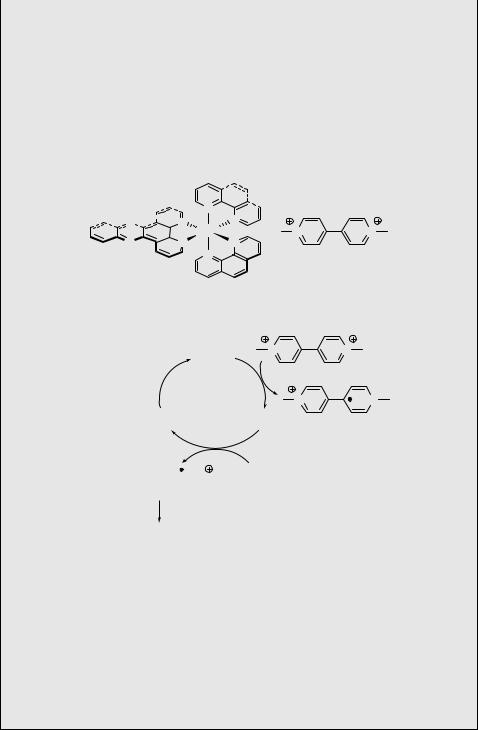
A light-driven shuttle is shown in the simplified Scheme 6.207.1246 The nanomachine 434 ( 5 nm in length) is composed of bis-p-phenylene-34-crown-10, an electron-rich macrocycle (M), which can move (shuttle) between two electronacceptor moieties ( stations ), 4,40-bipyridinium (A1) and 3,30-dimethyl-4,40- bipyridinium (A2), linked to the stoppers, the p-terphenyl moiety (S) and the tetraarylmethane group (T). The photoinduced shuttling movement starts with excitation of a [Ru(bpy)3]2 þ photosensitizer (P2 þ ), covalently attached to the molecule via S, following electron transfer to A12 þ to form A1 þ . As a result, the ring M moves to the adjacent A22 þ by Brownian motion, while a reverse electron transfer from A1 þ restores the charge on A12 þ , allowing M to move back. The ring-displacement process requires a time frame of 100 ms and operates with F 0.12.
Scheme 6.207
Case Study 6.31: Biochemistry – photocatalytic oxidation of DNA guanine
The photoexcited ruthenium(II) (phenanthroline)dipyridophenazine complex {[D-Ru (phen)2(dppz)]2 þ (435)}, an analogue of [Ru(bpy)3]2 þ (Scheme 6.204), can be oxidized
378 |
Chemistry of Excited Molecules |
by electron transfer to a weakly bound electron acceptor, such as methyl viologen [paraquat; N,N0-dimethyl-4,40-bipyridinium dichloride (436)] or [Co(NH3)5Cl]2 þ , resulting in the formation of a Ru3 þ intercalator (a molecule that binds to DNA and then inserts itself between base pairs) (Scheme 6.208).1247 This potent ground-state oxidant can be reduced back to the Ru2 þ complex by reaction with a reduced quencher or by electron transfer from a guanine base of DNA. The resulting guanine radical cation forms a neutral radical 437 by deprotonation, which can irreversibly react, for example, with molecular oxygen. This approach has been developed to explore electron transfer chemistry on double helical DNA (see also Special Topic 6.7).1248
|
|
|
N |
N |
|
|
|
N |
N |
Ru2+ |
N |
N |
|
N |
N |
N |
|
N |
|
|
|
|
|
|
|
|
|
[Ru(phen) (dppz)]2+ |
([RuL ]2+, 435) |
methyl viologen (436) |
|
|
2 |
3 |
|
|
|
|
|
|
[RuL ]2+* |
N |
N |
|
|
|
|
3 |
|
|
|
|
hν |
|
|
|
|
|
|
|
|
|
N |
N |
|
|
[RuL ]2+ |
|
[RuL ]3+ |
|
|
|
3 |
|
|
3 |
|
|
|
[guanine] |
+ H |
guanine (DNA) |
|
|
|
437 |
|
|
|
|
|
|
O2 |
|
|
|
|
oxidation products
Scheme 6.208
Experimental details.1247 DNA duplexes were formed from oligonucleotides and prepared on a DNA synthesizer by slow cooling of equal concentrations of complementary strands. Solutions of a duplex (8 mM), [Ru(phen)2(dppz)]2 þ (8 mM) and 10–20 equivalents of a quencher (e.g. methyl viologen), were irradiated at 436 nm with an Hg–Xe lamp (1000 W) equipped with a monochromator (Figure 3.28). After irradiation, the samples were treated with piperidine, dried and electrophoresed through polyacrylamide gel. The extent of DNA damage was evaluated by phosphorimaging.

Metalloporphyrins
In addition to [Ru(bpy)3]2 þ , many other metal complexes can be utilized in photoinduced redox reactions (see also Section 6.8). An important class of such compounds are metalloporphyrins, macrocycles derived from four pyrrole-like subunits with a coordinated metal (haemoglobin or cyanocobalamin (vitamin B12) are the best known derivatives). A free base porphyrin, and also some non transition metal (Al3 þ , Zn2 þ , etc.) complexes, are excellent and widely used photosensitizers1249 to generate singlet oxygen (Section 6.7.1). Many metalloporphyrins can also be involved in photoinduced electron transfer reactions; their redox properties depend on the nature of coordinated metal ion. Complexes of metals possessing a higher positive charge display higher standard potentials of oxidation.1248 Conjugation of the metal d-electrons with the p-electrons of the porphyrin macrocycle causes strong charge transfer and electrostatic interactions upon excitation (the absorption spectrum exhibits significant bands in the visible part of the spectrum). The central heavy metal ion also enhances intersystem crossing due to spin–orbit coupling (Section 4.8).
The interesting dual behaviour of porphyrin derivatives is demonstrated by intramolecular electron transfer between an electron-rich tris(4-methylphenyl)porphyrin zinc complex (ZnTPP) and an electron-deficient tris(4-methylphenyl)octafluoroporphyrin (TPOFP) (Teflon porphyrin) free base connected via an azobenzene moiety (438; Scheme 6.209).1250 Upon photolysis at >440 nm, E ! Z photoisomerization (Section 6.4.1) occurs. (Z)-438 can revert to (E)-438 only thermally, because the excited state of the Z-isomer is efficiently quenched by intramolecular electron transfer from ZnTPP to TPOFP, whereas no such interaction is possible in the E-isomer because the chromophores are too remote. This approach has been suggested for photocontrolled molecular electronics.
|
|
ZnTPP |
|
|
|
|
|
|
|
|
N |
ZnTPP |
|
|
N |
|
hν N |
|
|
|
(E)-438 |
N |
|
∆ |
|
electron |
|
|
|
|
|
|
transfer |
|
|
|
|
TPOFP |
|
|
|
|
TPOFP |
(Z)-438 |
|
|
|
|
|
|
|
|
R |
|
|
F |
R |
F |
|
|
|
|
F |
|
F |
|
N |
N |
|
|
N |
HN |
R |
Zn |
|
= ZnTPP; R |
|
= TPOFP |
|
N |
N |
|
|
NH |
N |
|
|
|
|
F |
|
F |
|
R |
|
|
F |
R |
F |
R = 4-methylphenyl |
|
|
|
|
380 |
Chemistry of Excited Molecules |
6.4.5Problems
1.Explain the following concepts and keywords: photochromism; photoswitching; photoinitiator; photoaffinity labelling; photo-Wolff rearrangement; photoactivatable compound; Barton reaction; photochemical trigger; photolabile linker; photoamination; redox photosensitization.
2.Suggest the mechanisms for the following reactions:
(a)
|
OAc |
OAc |
ONO |
O |
O |
|
OH |
H |
hν |
H |
H |
H |
H |
O |
|
O |
|
|
HO N |
[ref. 1251]
(b)
[ref. 1252]
(c)
O |
hν |
O |
|
|
O |
|
|
|
|
|
|
|
|
|
|
(>320 nm) |
|
H |
|
H |
SiMe3 |
CN |
|
+ |
|
|
H |
|
|
N |
N |
|
N |
H |
|
|
|
|
|
|
|
|
CN |
|
|
|
|
[ref. 1253]
3.Predict the major photoproduct(s):
(a)
N2
hν
O
N
(b)
[ref. 1255]
(c)
C8H17
hν
MeOH
N
OH
[ref. 1256]
6.5 Sulfur Compounds
Sulfur and oxygen (see Section 6.3) compounds are isoelectronic and the discussion of their excited states is analogous. The differences can be understood by recalling two simple facts: sulfur is less electronegative than oxygen and the C¼S p-bond is weaker than the C¼O p-bond. The absorption spectra of thiophenols and thioethers are similar to those of the corresponding phenols (Figure 6.15, top).
Figure 6.15 Absorption spectra of sulfur-containing compounds. Top: thiophenol (in heptane, —), thioxanthone (in hexane, ), thiourea (in water, - - -), bis(trifluoromethyl)thioketene (in
isooctane, . . . . ).280 Bottom: absorption (—) and emission (. . . . ) spectra of adamantanethione in perfluoroalkane solvents.1257 The peak at the red edge of the S0 ! S1 transition coincides with
the 0–0 band of phosphorescence and is attributed to S0 ! T1 absorption
382 |
|
|
|
Chemistry of Excited Molecules |
|
|
|
|
|
H |
H |
MeS |
S |
H2N |
|
|
EtO |
|
|
S |
|
|
S |
|
|
S |
|
|
|
|
S |
|
|
|
S |
|
|
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
Me |
Me |
S |
Me |
|
|
Me |
1.64 |
1.79 |
2.24 |
2.14 |
|
2.72 |
|
|
2.65 |
|
Br |
Cl |
F |
F3C |
Me Si |
|
|
H N |
|
|
S |
|
|
S |
|
|
S |
|
|
|
S |
3 |
|
|
S |
2 |
|
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Br |
Cl |
F |
F3C |
Me Si |
|
|
H N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
|
|
2 |
|
|
2.08 |
2.08 |
2.84 |
1.72 |
|
|
1.89 |
|
|
>4.0 |
|
Figure 6.16 Positions of the n,p transitions ( ~v/mm 1) in thiocarbonyl compounds
The n,p transitions of thiocarbonyl compounds lie at much longer wavelengths ( 500 nm) than those of the corresponding carbonyl compounds, because its np lone pair lies at much higher energy. The energy gap between the S1(n,p ) and S2(p,p ) states of thiocarbonyl compounds is therefore fairly large and the Kasha and Vavilov rules (Section 2.1.8) may be violated (Figure 6.15, bottom). Even inefficient reactions proceeding from the S2(p,p ) state can amount to the dominant photoprocess because the S1(n,p ) states are short-lived (energy gap law) and not very reactive due to their low excitation energy. Spin–orbit coupling between the ground state and the lowest triplet state of n,p character is sufficiently large in thiocarbonyl compounds to render singlet–triplet absorption detectable (« 1 M 1 cm 1; Figure 6.15, bottom).
The positions of the n,p transitions in a series of thiocarbonyl compounds are shown in Figure 6.16.1258 The shifts between related compounds follow the trends expected from perturbation theory: inductive acceptors lower the energy of the np orbital on sulfur and mesomeric interactions raise the energy of the p orbital.
Table 6.17 Photoprocesses involving excited sulfur-containing compounds
Entry |
Starting materiala |
Product(s) |
|
Mechanism |
Section |
|
|
|
|
|
|
|
|
|
|
|
S |
* |
|
|
|
SH |
|
|
|
1 |
[H] |
|
|
Photoreduction/Photoaddition |
6.5.1 |
|
+ |
|
|
|
S |
* |
|
|
|
H |
|
|
|
2 |
+ |
[H] |
S |
|
Photoreduction/Photoaddition |
6.5.1 |
|
|
|
|
S |
* + |
|
|
|
S |
|
|
|
3 |
|
|
|
|
|
|
Photocycloaddition |
6.5.1 |
4 |
R R' |
* |
|
|
|
R S |
R' |
Photofragmentation |
6.5.2 |
S |
|
|
|
|
O O |
|
|
O O |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a[H] ¼ hydrogen atom donor.
Similarly to carbonyl compounds (Section 6.3.1), thiocarbonyl compounds abstract hydrogen upon irradiation; however, both n,p and p,p excited states are reactive and the hydrogen atom can be added to either the sulfur (Table 6.17, entry 1) or carbon (entry 2) atoms of the C¼S bond. Aliphatic and aromatic thiocarbonyl compounds can also undergo photocycloaddition to unsaturated compounds from both singlet or triplet excited states to form thietanes (analogously to the Paterno`–B€uchi reaction; see Section 6.3.2) (entry 3) or 1,4-dithianes. On the other hand, fragmentation of the S–C bond is a typical primary process observed in excited sulfones and sulfonates (entry 4), followed by efficient SO2 extrusion from the radical intermediate.
6.5.1Thiocarbonyl Compounds: Hydrogen Abstraction and Cycloaddition
S |
SH |
S |
hν , [H] |
S |
|
|
|
H |
|
|
|
|
R R |
R R |
R R |
|
R R |
Recommended review articles.1259–1264
Selected theoretical and computational photochemistry references.1258,1265–1268
The differences in the photoreactivity of carbonyl and thiocarbonyl compounds are
generally linked to the excitation energies (compare: benzophenone ES ¼ 316 kJ mol 1, ET ¼ 288 kJ mol 1; thiobenzophenone ES ¼ 191 kJ mol 1, ET ¼ 165 kJ mol 1),157 a high
polarizability of the C¼S bond and the softer character and lower electronegativity of sulfur compared with oxygen (Section 6.3).1260,1262,1268 Thiones in solution are known to
undergo hydrogen abstraction, cycloaddition, oxidation and fragmentation reactions from both their singlet and excited triplet states.1261
Hydrogen Abstraction by the Excited Thiocarbonyl Group
In contrast to excited carbonyl compounds, which abstract hydrogen efficiently from the n,p state (Section 6.3.1), thiocarbonyl compounds react from both n,p and p,p states
and the hydrogen atom can be added to either sulfur or carbon atom of the C¼S bond.1259,1269,1270 The sulfur atom is larger than oxygen, therefore the reaction can occur
over greater C¼S :H distances than those involving the carbonyl group.1271 Singlet p,p excited (S2) alkyl aryl thioketone 439, for example, undergoes unexpected photoinduced intramolecular 1,6-hydrogen atom abstraction to form a short-lived 1,5-biradical intermediate, which cyclizes to the cyclopentanethiol 440 exclusively, although with a low quantum yield (F ¼ 0.06) (Scheme 6.210).1272 The hydrogen abstraction is competitive with back hydrogen transfer and excited state decay. A Norrish type II 1,4-biradical intermediate and subsequent fragmentation yielding elimination products typical of the triplet n,p excited alkyl aryl ketones are not observed here.
In contrast, hydrogen atom transfer to carbon of the C¼S bond occurs in excited (both n,p and p,p ) 2,4,6-tri-tert-butylthiobenzaldehyde (441), resulting in the six-membered thiolane derivative 442 formation via a radical mechanism (Scheme 6.211) in nearly quantitative chemical yield.1273
384 |
Chemistry of Excited Molecules |
S |
|
1 |
S |
* |
hν |
|
|
|
|
(S2) |
Ph |
|
|
Ph |
|
|
|
439 |
|
|
1,6-hydrogen |
|
|
|
|
|
|
|
|
transfer |
HS |
|
|
HS |
|
|
|
Ph |
|
Ph |
|
|
|
440 |
|
|
|
|
Scheme 6.210 |
|
H S |
|
H |
S |
S |
|
H |
|
|
|
|
hν
Scheme 6.211
Case Study 6.32: Mechanistic photochemistry – reactions from different excited states
Excitation of the alkyl aryl thione 443 with an activated b-hydrogen into either the S1 (n,p ) or S2 (p,p ) state using different irradiation wavelengths was found to produce the cyclopropanethiol 444 via the same 1,3-biradical (Scheme 6.212).1274 In contrast to the reaction from the 1p,p state, the product formation obtained by excitation into S1 was quenched by a triplet quencher, indicating that the lowest (n,p ) triplet must be the reactive state. A very large isotope effect (kH/kD > 17) found for this reaction indicates a substantial tunnelling effect (Special Topic 5.2) in the hydrogen abstraction process.
S Ph |
SH Ph |
Ph |
|
|
HS |
|
hν |
|
443 |
|
444 |

Experimental details.1274 A benzene solution of 443 (5 10 3 M) was irradiated using a medium-pressure mercury lamp in a Pyrex immersion well through an optical filter (lirr > 420 nm; S1 excitation; Figure 3.9) to complete conversion. Preparative TLC and crystallization from n-pentane afforded the product in 88% chemical yield.
The same benzene solution was irradiated through a Pyrex filter (lirr > 280 nm; S2 excitation) to complete conversion and the product was isolated in 82% chemical yield.
Photocycloaddition to Alkenes
Both aliphatic and aromatic thiocarbonyl compounds can undergo photocycloaddition to unsaturated compounds from either singlet or triplet excited states.1260,1261,1263 The
intersystem crossing quantum yields in aromatic thioketones are usually high and are dependent on the excitation wavelength: F is less than unity ( 0.5) in the case of
excitation to the second excited singlet state, S2 (lexc 300–400 nm), but it approaches F ¼ 1 when the irradiation is done at lexc > 500 nm (S1).1275 Excited thiobenzophenone
(445), for example, reacts with an electron-rich enol ether 446 to produce the thietane 447, analogous to the Paterno`–B€uchi reaction involving carbonyl compounds (Section 6.3.2) or the 1,4-dithiane 448, formed by the reaction of two molecules of thiobenzophenone.1276 Whereas both products are obtained from the n,p excited triplet state (T1) of thiobenzophenone and electron-rich ethoxyethene via a radical mechanism, the thietane 449 is formed exclusively by a stereospecific cycloaddition from the p,p excited singlet state (S2) to an electron-deficient trans-fumaronitrile (450) (Scheme 6.213).
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
S |
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
EtO |
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
|
|
|
hν |
|
|
|
S |
|
|
|
S |
Ph |
+ |
|
|
|
|
|
|
|
+ |
|
|
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(via T1) |
|
|
|
|
|
|
|
|
|
|
|
H |
|
|
|
Ph |
|
|
|
|
Ph |
Ph |
EtO |
|
|
|
|
H |
|
S |
|
Ph |
|
|
|
|
|
|
|
|
|
|
|
EtO |
|
|
|
Ph |
|
EtO |
Ph |
445 |
|
446 |
|
|
|
|
|
447 |
|
448 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CN |
|
H |
|
CN |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
S |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
445 |
+ |
|
|
|
|
hν |
|
NC |
|
|
|
|
|
(via S2) |
|
|
|
|
|
|
|
|
H |
|
|
|
|
NC |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Ph Ph |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
450 |
|
|
|
|
|
|
|
449 |
|
|
|
|
|
|
|