396 |
Chemistry of Excited Molecules |
Alkyl Halides
Simple alkyl chlorides and bromides do not absorb above 250 nm, whereas the absorption band of an n,s transition in methyl iodide, for example, has a maximum at lmax ¼ 258 nm.1334 Direct irradiation of alkyl halides results in homolytic cleavage of the C X bond (X ¼ halogen). In general, the excitation energy (ES) in alkyl iodides or
bromides (380–475 kJ mol 1) is higher than the C X bond dissociation energies (220–300 kJ mol 1).1314 The quantum yield of the fragmentation does not have to be
unity even in the gas phase because of possible concurrent non-radiative or radiative energy dissipation processes.1335 The photoreaction usually involves the excited singlet state and reaction is initiated by homolytic cleavage, possibly followed by electron transfer within the caged radical pair (Scheme 6.228). The radical species undergo radical reactions, such as hydrogen abstraction or rearrangement, while the resulting carbocation can be attacked by nucleophiles (nucleophilic photosubstitution; SN1 ).
R X |
hν |
X |
[H] |
R |
RH |
|
homolytic |
|
hydrogen |
|
cleavage |
|
abstraction |
|
|
|
electron |
|
|
|
transfer |
|
R |
X |
Nu |
|
|
|
|
|
nucleophilic |
|
|
|
attack |
Scheme 6.228
Irradiation of primary alkyl bromides or iodides affords typically a mixture of reduction and elimination products. For example, a radical pair produced by photolysis of the halooctanes 469 undergoes in the initial stage either competing diffusion and hydrogen abstraction from the solvent molecules (to give 470) or electron transfer giving rise to elimination product (471) – a favoured process in case of alkyl iodides (Scheme 6.229).1336 Formation of the nucleophilic substitution product 472 is not observed, because solvent does not participate in carbocation generation.
|
hν |
|
|
n-C7H15CH2X |
|
n-C7H15CH3 |
+ n-C6H13CH=CH2 |
+ n-C7H15CH2OMe |
|
|
MeOH |
|
|
469 |
470 |
471 |
472 |
X = Br |
66% |
31% |
not detected |
X = I |
38% |
56% |
not detected |
Scheme 6.229
Photolysis of norbornan-1-yl (tertiary) halides (473) in methanol affords both reduction and the nucleophilic photosubstitution products 474 and 475, respectively (Scheme 6.230).1336
Since no detectable incorporation of deuterium was observed to occur in CH3OD, 475 cannot be produced via an elimination process to give a bridgehead alkene (bicyclo[2.2.1]hept-1-ene, 476), followed by acid-catalysed addition of an alcohol. The ionic (electron transfer) mechanism is apparently preferred again in the photolysis of iodides, although the interplay between ionic versus radical mechanism is also dependent on the ionization potentials of the alkyl radicals (that subsequently form carbocation intermediates). For example, an electronwithdrawing hydroxy group in position 3 of 473 stimulates formation of the corresponding
reduction product because the ionization potential of the 3-hydroxynorbornan-1-yl radical is higher than that of norbornan-1-yl radical.1337
|
|
hν |
+ |
|
|
MeOD |
|
X |
MeO |
|
D |
|
473 |
474 |
475 |
|
X = Br |
55% |
30% |
|
X = I |
12% |
76% |
|
hν |
|
|
|
|
MeOD |
|
|
476 |
MeO D |
|
Scheme 6.230
Photochemistry of polyhaloalkanes results in a C X bond fission of the weakest bond in the initial step. For example, irradiation of bromotrichloromethane affords trichloromethyl
and bromine radicals, which add to styrene to obtain 1-bromo-3,3,3-trichloropropylben- zene (477) in 78% chemical yield (Scheme 6.231).1338
|
|
Br |
Cl3CBr |
hν |
Br |
Cl3C |
|
|
CCl3 |
|
|
477 |
Scheme 6.231
Vinyl Halides
Photolysis of vinyl iodides is a convenient method for the generation of vinyl cations, highly reactive intermediates that are difficult to generate by thermal processes. Irradiation of 1-iodocyclohexene (478, X ¼ I) in methanol in the presence of zinc as an iodine

398 |
Chemistry of Excited Molecules |
scavenger affords the major nucleophilic substitution product 479 and small amounts of the radical-derived reduction product cyclohexene (480) (Scheme 6.232).1339 In the absence of zinc, the ketal 481 is apparently obtained via an acid-catalysed addition of methanol to 479. In contrast, vinyl bromide (478, X ¼ Br) mainly provides the photoreduction product 480, which parallels closely the photobehaviour of the saturated analogues.1314
|
X |
X |
|
|
X |
|
|
|
hν |
|
|
478 |
[H] |
MeOH |
|
|
|
|
|
OMe |
OMe |
|
|
OMe |
|
|
H |
|
|
MeOH |
|
|
480 |
479 |
481 |
X = Br |
62% |
|
26% |
X = I |
20% |
|
72% |
Scheme 6.232
Aryl Halides
Simple aromatic halides display two principal superimposing absorption bands, p,p and s,s , tailing over 250 nm. Intersystem crossing (ISC) to T1 can be fast and efficient due to
increased spin–orbit coupling attributed to the heavy-atom effect (Section 4.8) of halogen atoms (Br, I).842,849,1316 The subsequent C X bond homolysis may afford aryl radicals and
halogen atoms as the primary products (Scheme 6.233), which requires that the energy of the reactive excited state be greater than the corresponding C X dissociation energy. Fission is, therefore, thermodynamically unfavourable for aryl fluorides, for example
fluorobenzene (ET ¼ 353 kJ mol 1; DC F ¼ 526 kJ mol 1), and for most aryl chlorides, for example 1-chloronaphthalene (ET ¼ 248 kJ mol 1; DC Cl ¼ 403 kJ mol 1).849 In the latter
|
|
|
|
|
|
|
|
Ar-X |
hν |
1 [ Ar-X ]* |
|
1 [ Ar + X ] |
|
|
X = Cl, Br, I |
ISC |
|
homolysis |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3 [ Ar-X ]* |
|
3 [ Ar + X ] |
|
|
|
|
|
|
|
electron |
|
|
|
|
|
|
transfer |
|
|
|
|
|
|
|
|
|
|
|
|
3 |
[ Ar + X ] |
case, inefficient photodegradation may take place from the singlet state.1340 Triplet state homolysis can be exothermic in iodoand bromoarenes. The resulting triplet radical pair can be involved in subsequent radical reactions or undergo in-cage electron transfer to form an aryl cation, which is readily attacked by nucleophiles such as water.
Photodehalogenation (photoreduction) – replacement of the halogen atom by hydrogen – in the presence of hydrogen-atom donors is an interesting alternative to Raney nickelcatalysed reduction. Irradiation of 2-bromophenol (482) in propan-2-ol, for example, leads to phenol in >90% chemical yield via a radical mechanism (Scheme 6.234).1341
OH OH
Br hν
- Br
482
Scheme 6.234
There is much evidence that photodehalogenation reactions of triplet excited halogenated biphenyls (483), naphthalenes and other aromatic compounds (which exhibit efficient intersystem crossing) in the presence of good electron donors, such as amines, is
accompanied by full or partial electron transfer to form exciplex intermediates (Scheme 6.235).667,849 Escape of a free radical ion 484 may give the corresponding
hydrocarbon in the presence of a proton donor such as water or a good hydrogen-atom donor ([H]).
|
3[ Ar-X ]* |
|
R3N |
|
δ |
|
|
|
δ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
[ Ar-X |
NR3]* |
|
exciplex |
|
483 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
formation |
|
|
|
|
- R3N |
|
|
|
[H] |
|
|
|
ArX |
|
|
|
|
[H ] |
|
ArH |
|
|
Ar + X |
|
|
|
|
|
|
|
|
ArH + X |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
484 |
|
|
|
|
|
Scheme 6.235
The synthetically useful ring closure reaction (intramolecular cyclization) of 1-(o-chloropheny1)naphthalene (485) gives fluoranthene (486) in 72% chemical yield (Scheme 6.236).1342 This reaction proceeds via initial homolysis of the C Cl bond, followed by radical aromatic substitution.
Some important persistent environmental pollutants, such as 1,1,1-trichloro-2,2-bis (4-chlorophenyl)ethane (DDT), polychlorinated biphenyls (PCBs), dibenzo-p-dioxins (PCDDs) and polybrominated diphenyl ethers (PBDEs), are aryl halides; therefore, studies
400 |
Chemistry of Excited Molecules |
Cl |
hν |
H |
|
|
|
- H |
|
- Cl |
485 |
|
|
486 |
|
|
Scheme 6.236 |
|
of the photochemistry of halogenated aromatic compounds have also been stimulated by environmental concerns.849 Photolysis of chlorinated biphenyls in non-nucleophilic
media, for example, exhibits a preferential dehalogenation of the o-chlorine atoms, because their loss relieves the steric strain (Scheme 6.237).849,1343
hν
φ = 0.39
Cl
hν
Cl φ = 0.001
Scheme 6.237
In aqueous solutions, 2-halophenols, such as 2-bromophenol, are known to undergo photo-Wolff rearrangement (see also Scheme 6.171). In this reaction, the cyclopentadienecarboxylic acid derivatives 487 are formed via the singlet a-ketocarbene (488)
and ketene (489) intermediates, followed by nucleophilic addition of water (Scheme 6.238).1344–1346
1 OH |
* |
O |
O |
|
C |
|
|
Br |
|
H2O |
|
|
|
|
|
|
|
|
|
- HBr |
|
COOH |
|
|
|
|
|
|
488 |
489 |
487 |
Scheme 6.238
Irradiation of 3,4-difluoroacetophenone (490) in acidic aqueous solutions leads to an unusual product of solvolysis, 4-fluoro-3-hydroxyacetophenone (491), in nearly quantitative chemical yield (Scheme 6.239).332 Nucleophilic addition of water to the meta-position proceeds via O-protonated triplet acetophenone.
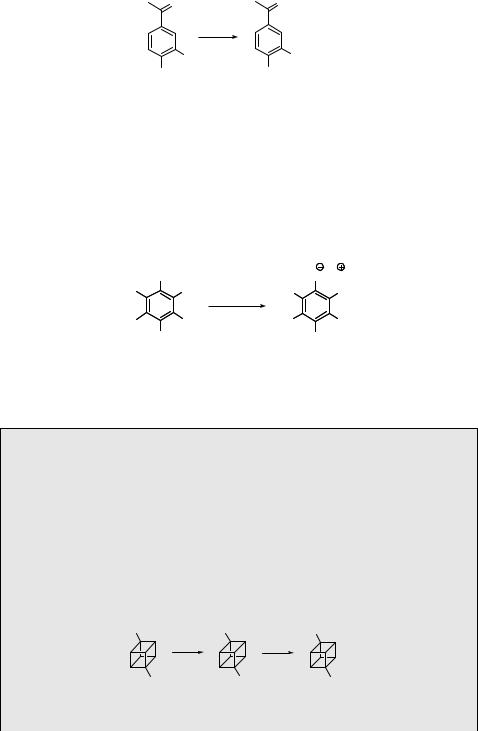
Halogen Compounds |
401 |
O |
O |
|
|
hν |
+ HF |
|
H O, H+ |
|
|
F |
2 |
OH |
|
F |
F |
|
490 |
491 |
|
Scheme 6.239
Nucleophilic photosubstitutions are typical reactions of aromatic halides. The variability of the process in terms of mechanism is presented in detail, along with many examples, in Section 6.2.3; therefore only one example of the reaction is mentioned here. Multiple photocyanation of hexachlorobenzene (492) takes place in aqueous acetonitrile containing a cyanide ion to give pentacyanophenolate (493) (Scheme 6.240).1347 The reaction involves the triplet state apparently via an SN2Ar (Scheme 6.93) pathway.
Cl |
|
|
O Na |
Cl |
Cl |
NC |
CN |
|
|
hν, NaCN |
|
Cl |
Cl |
MeCN, H2O NC |
CN |
Cl |
|
|
CN |
492 |
|
|
493 |
Scheme 6.240
Case Study 6.35: Synthesis of cage compounds – cubane
Photolysis of 1,4-diiodocubane (494) in nucleophilic solvents, such as methanol, was found to provide effectively the substitution product 495 (Scheme 6.241).1348 It was argued that the reaction proceeds via the formation of a caged radical pair and electron transfer to give iodide ion and a carbocation, which is trapped by the solvent (SN1 pathway). Hyperconjugative stabilization in cubyl cation cannot assist much from an extremely strained olefin cubene. However, the authors expected that photochemical fission of the C I bond would still provide sufficient energy for production of the cation intermediate.
I |
I |
MeO |
hν |
|
hν |
MeOH |
|
MeOH |
I |
OMe |
OMe |
494 |
|
495 |
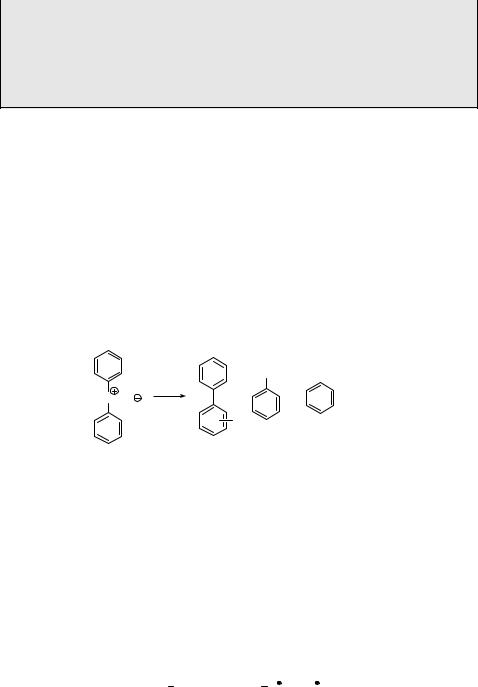
402 |
Chemistry of Excited Molecules |
Experimental details.1348 1,4-Diiodocubane (494; 0.2 mmol) suspended in dry methanol (6.0 ml) in a quartz tube equipped with a magnetic stirrer was irradiated in a Rayonet reactor equipped with 16 low-pressure mercury lamps (lirr ¼ 254 nm) (Figure 3.10) under a nitrogen atmosphere at 40 C. About 32% monosubstituted 1-iodo-4-methoxycubane was detected by GC after 6 h. Further irradiation gave a significant production of l,4-dimethoxycubane (495; 50%).
Hypervalent Iodine Compounds
Hypervalent iodine compounds, especially organoiodine(III) species, are commonly used in synthetic organic chemistry, but photochemical applications are still rare.1349 Direct photolysis of diphenyliodonium triflate (496), for example, affords a singlet excited species that undergoes a heterolytic cleavage leading to phenyl cation and iodobenzene. In addition, intersystem crossing leads to the triplet state, which is homolytically fragmented to give a caged pair of iodobenzene radical cation and phenyl radical. The primary isolated products in H-atom donating solvents are 2-, 3- and 4-iodobiphenyl, iodobenzene, benzene and trifluoromethanesulfonic acid (Scheme 6.242).1350 Such a photochemically generated strong acid has been successfully used as an initiator of cationic polymerizations1351 (Section 6.8.1).
|
|
I |
|
I |
hν |
+ |
+ TfOH |
+ |
TfO |
[H] |
|
|
|
I |
|
|
496
Scheme 6.242
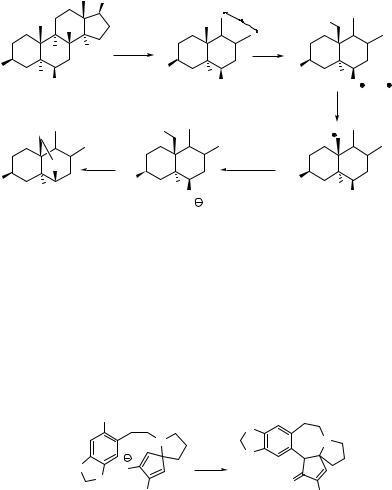
Alkyl Hypohalites
Hypohalites represent another class of photolabile organic halogen compounds, which exhibit two absorption peaks above 250 nm; alkyl hypochlorites have lmax at 260 and320 nm, while the absorption of alkyl hypobromites and hypoiodites is bathochromically shifted.1319 Irradiation of alkyl hypohalites leads to a homolytic cleavage to give alkoxyl and halogen radicals with a high photochemical efficiency (Scheme 6.243).1352 These primary intermediates can subsequently undergo various reactions, such as hydrogen abstraction, rearrangement or fragmentation.
hν
R OX  R O + X
R O + X
Alkyl hypochlorites and hypobromites are relatively stable molecules; alkyl hypoiodites can only be prepared in situ, usually by the reaction of alcohols with metal acetates or oxides and iodine or by the reaction of alcohols with a hypervalent iodine compound and iodine.1319 Alkyl hypochlorites and hypoiodites can be utilized in reactions that parallel those of the organic nitrites (Barton reaction; Section 6.4.2). For example, the photochemistry of the steroidal hypoiodite 497, prepared by the reaction of the corresponding alcohol with iodine oxide (I2O) generated from mercury(II) oxide and molecular iodine in situ, affords a new five-membered ring via an O–I bond fission, 1,5- hydrogen abstraction and substitution (Scheme 6.244).1353
|
C8H17 |
|
|
H |
|
|
H |
|
|
|
H |
HgO, I2 |
hν |
|
H |
|
|
AcO |
AcO |
H OI |
AcO |
H OH |
|
H O + I |
|
|
497 |
|
|
I |
|
|
O |
|
I2 or ROI |
|
AcO |
AcO |
|
AcO |
H |
H |
|
H |
|
O |
|
OH |
Scheme 6.244
6.6.3Problems
1.Explain the following concepts and keywords: photoinduced radical chain mechanism; photonitrosylation; nucleophilic photosubstitution; photodehalogenation.
2.Suggest the mechanisms for the following reactions:
(a)
|
I |
|
O |
|
N |
|
|
|
N |
|
|
hν |
O |
O |
O |
|
|
|
|
O |
|
O |
|
MeO |
|
OMe |
|
404 |
Chemistry of Excited Molecules |
|
|
|
(b) |
|
|
|
|
|
|
|
hν, NBS |
N |
N |
N |
N |
|
N |
O |
|
+ |
|
HN |
NH |
N |
N |
|
N |
|
H |
|
O |
O |
|
O |
|
|
|
|
|
NBS = N-bromosuccinimide
[ref. 1355]
3.Predict the major photoproduct(s):
(a)
hν
OCl
[ref. 1356]
(b)
Br
O
hν
+ CH2NO2
NH3 (l)
[ref. 1357]
6.7 Molecular Oxygen
The photophysical properties of oxygen were discussed in Section 2.2.5. Molecular oxygen can be involved in photoreactions as either the ground-state (triplet;3O2) or an excited (singlet;1O2) species. Section 6.7.1 discusses the oxidation reactions of excited organic compounds with ground-state oxygen, called oxygenations (Table 6.20, entry 1). When a triplet sensitizer, such as Methylene Blue, Rose Bengal or porphyrin, is introduced, its excitation is readily transferred to ground-state oxygen to form 1O2 (entry 2). Singlet oxygen can also be formed by thermal decomposition of unstable molecules, such as endoperoxides (entry 3).
1O2 is a reactive, short-lived molecule, which can undergo several specific reactions with organic unsaturated hydrocarbons. Oxygenation of a carbon–carbon double bond to give 1,2-endoperoxides (entry 4) can be formally interpreted as [2 þ 2] cycloaddition, whereas addition of singlet oxygen to a 1,3-diene moiety, such as a buta-1,3-diene derivative or benzene ring, to form 1,4-endoperoxides (entry 5) correspond to [2 þ 4] cycloaddition. Endoperoxides may be further photolysed to rearrange to diepoxides (entry 6) or to revert back to the corresponding diene and oxygen molecule. When an alkene reactant contains at least one allylic hydrogen, the ene reaction is another competing process (entry 7).
|
Molecular Oxygen |
405 |
Table 6.20 Photoprocesses involving molecular oxygen |
|
|
|
|
|
Entry Starting material |
Product(s) |
Mechanism |
Section |
1 R* þ 3O2
3 O
O
4 + 1O2
+ 1O2
5 + 1O2
+ 1O2
O
Oxidation products |
Oxygenation with ground state |
6.7.1 |
|
|
|
|
|
|
oxygen |
|
1O2 þ sensitizer |
Singlet oxygen production |
6.7.1 |
|
|
|
|
|
+ 1O2 |
Singlet oxygen production |
6.7.1 |
|
O |
|
O |
[2 þ 2] photooxygenation |
6.7.2 |
|
|
|
|
|
|
|
|
|
|
O O |
[4 þ 2] photooxygenation |
6.7.2 |
|
|
|
|
O |
|
|
|
|
O |
Endoperoxide |
6.7.2 |
|
|
|
|
|
|
|
|
|
|
|
|
photorearrangement |
|
HOO |
|
|
|
|
|
|
|
|
Ene reaction |
6.7.3 |
|
|
|
|
|
|
|
|
CH2
6.7.1Molecular Oxygen: Ground State and Excited State
|
hν |
3O2 |
|
|
|
|
|
|
|
R |
|
R* |
|
|
|
|
products |
3O2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
O O |
∆ |
|
+ 1O2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Recommended review articles.135,136,1358–1363
Selected theoretical and computational photochemistry references.135,136,1364
Ground-state Molecular Oxygen
Ground-state molecular oxygen (dioxygen), an open-shell triplet (biradical) denoted by spectroscopists 3Sg, is one of the most abundant and important life-supporting species on Earth. It can participate in some photochemically initiated reactions, sometimes called