

Photochemistry_of_Organic
.pdf
340 |
Chemistry of Excited Molecules |
6.4 Nitrogen Compounds
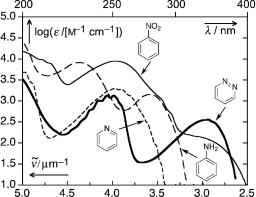
The absorption spectra of some nitrogen-containing benzene derivatives are shown in Figure 6.8. The lowest excited singlet state of aromatic nitro compounds is of n,p character. Aza substitution in the ring, as in pyridine or quinolines, does not shift the absorption bands much with respect to those in the parent hydrocarbons benzene and naphthalene, respectively, but enhances the intensity of the 1Lb bands that now reach «max 103 M 1 cm 1 (see Section 4.7). Moreover, the presence of low-lying n,p bands, where n corresponds to the lone pair orbital of the aza nitrogen, strongly influences the photophysical properties. The weak n,p bands are usually detectable only as shoulders on the red edge of the first p,p band. In 1,2-diaza compounds (pyridazine), lone-pair interaction raises the energy of the antisymmetric lone pair orbital. The first n,p band is then stronger (symmetry allowed) and well separated from the p,p absorption.
Figure 6.8 Absorption spectra of some benzene derivatives: nitrobenzene (––), aniline (– – –), pyridine (---), pyridazine ( ).280
The absorption spectra of pyrrole and indole (not shown), in which the nitrogen atom contributes two pz-electrons to the p-system, are also related to those of the isoelectronic hydrocarbons benzene and naphthalene, respectively. p-Donation is somewhat weaker for exocyclic amines such as aniline (Figure 6.8), which is not completely planar in the ground state. Aniline is a weak base; the pKa of its conjugate acid is 4.6. Protonation in acidic solutions converts the amino group to an inductive acceptor; the absorption spectra of protonated anilines are then similar to those of the corresponding benzene derivatives.
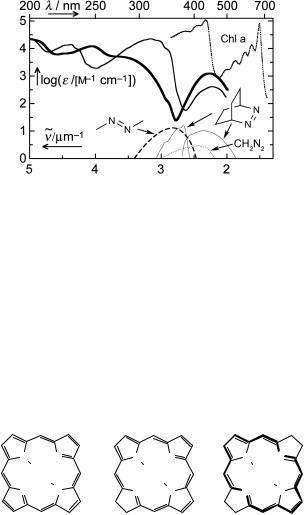
Azoalkanes exhibit an n,p absorption band in the near-UV region (Figure 6.9). The
n,p transition of the E-isomers is symmetry forbidden, log(«/[M 1 cm 1]) 1; that of the Z-isomers is allowed, log(«/[M 1 cm 1]) 2.1057 The n,p transitions are more intense in
the two azobenzene isomers and shifted to longer wavelengths, lmax ¼ 450 nm. Electrondonating substituents such as a p-dimethylamino group strongly shift the p,p transitions to longer wavelengths. Azo compounds undergo very rapid E–Z isomerization from the lowest n,p -excited singlet state; hence fluorescence and ISC quantum yields are usually very low. When E–Z isomerization of azo compounds is inhibited by steric constraints, fluorescence, nitrogen elimination or ISC become competitive (see below).
Nitrogen Compounds |
341 |
Figure 6.9 Absorption spectra of azobenzene (hexane, E-isomer —, Z-isomer ),280 azomethane (hexane, E-isomer, – – –),280 chlorophyll a (Chl a; in diethyl ether, . . . .),280 diazomethane (gas phase, ....);1058 absorption and fluorescence spectra of 2,3-diazabicyclo [2.2.2]oct-2-ene [in perfluoromethylcyclohexane, — (2 thin lines); shifted downward by 1.5 units for better visibility]1059
Many important pigments and dyes occurring in nature or used in technical applications are based on the cyclic tetrapyrrole chromophore porphyrin (Figure 6.10). There are 24 atoms participating in the conjugated system, but two double bonds (carbons 2, 3, 12 and 13) are easily hydrogenated, leaving a conjugated 18-membered ring that is bridged by two pyrrole-type nitrogen atoms (bacteriochlorin). Whereas chlorophylls (Figure 6.9) are 2,3-dihydroporphyrin (chlorin) derivatives having a magnesium ion replacing the two central hydrogen atoms, bacteriochlorophylls are 2,3,12,13-tetrahydroporphyrin (bacteriochlorin) derivatives (18-atom conjugated ring).
|
|
12 |
|
|
|
|
|
13 |
|
|
|
N |
N |
N |
N |
N |
N |
|
H |
|
H |
|
H |
|
H |
|
H |
|
H |
N |
N |
N |
N |
N |
N |
3 |
|
|
|
|
|
2 |
|
|
|
|
|
porphyrin |
chlorin |
bacteriochlorin |
|||
Figure 6.10 Porphyrin and its analogues
All porphyrins exhibit two types of absorption bands, the so-called Q-band(s) of moderate intensity in the visible region and a very strong band in the near-UV, the B-band or Soret band. Such a pattern is predicted by simple MO models for cyclic systems with 18 p-electrons (Section 4.7, Figures 4.28 and 4.29). In the highly symmetric zinc tetrakis (perfluorophenyl)porphyrin (TFPP), the symmetry-forbidden Q-band lies at 578 nm,
log(«/[M 1 cm 1]) ¼ 3.7, with a vibrational progression at 543 nm; the symmetry-allowed B-band at 412 nm is much more intense, log(«/[M 1 cm 1]) ¼ 5.7.1060 Both the Q- and
B-bands are due to a degenerate excited state in molecules that belong to the D4h point group. When the symmetry is lowered as in the metal-free H2(TFPP), the Q-band splits

342 |
Chemistry of Excited Molecules |
into two, Qx ¼ 635 and 582 nm and Qy ¼ 535 and 505 nm. Figure 6.9 shows the B- and Q- bands for chlorophyll a, a magnesium chlorin derivative.
The great structural diversity of nitrogen-containing compounds, ranging from imines and azo compounds to nitro compounds and amines, is connected to its diverse but characteristic photochemical reactivity (Table 6.15). The presence of a lone electron pair on nitrogen in chromophores containing an N¼X bond (X¼N, C) indicates that both the n,p and p,p excited states can be involved in the reactions. While E–Z isomerization is a typical reaction for both imines and azo compounds (entry 1), the latter chromophores may additionally
Table 6.15 Examples of primary photoprocesses of excited nitrogen-containing compouunds
Entry Starting materiala Product(s) Mechanism Section
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R' |
* |
|
N |
|
|
X |
|
|
|
|||||||||||||
1 |
|
|
N |
|
|
|
|
|
X |
|
|
|
|
|
E–Z isomerization |
6.4.1 |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
R |
|
|
|
|
|
|
|
|
|
R' |
|
|||||||||||||||||||
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
X = C, N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R' |
* |
R |
|
|
|
|
|
+ N2 |
+ R' |
|
|
||||||||||
2 |
|
|
N |
|
|
N |
|
|
|
|
|
|
Photofragmentation/ |
6.4.2 |
||||||||||||||||||||||||
|
|
|
||||||||||||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||
|
|
R |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
photoelimination |
|
|||||
3 |
|
R C |
|
|
N |
|
|
|
|
|
* |
R2C |
+ |
N2 |
Photofragmentation/ |
6.4.2 |
||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||
|
2 |
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
photoelimination |
|
||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
R |
|
|
|
|
|
|
+ N2 |
|
|
||||
4 |
|
R |
|
|
|
|
|
N |
|
|
|
|
|
|
|
|
|
|
|
Photofragmentation/ |
6.4.2 |
|||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
photoelimination |
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
R |
|
|
|
|
|
N |
|
+ N2 |
Photofragmentation/ |
6.4.2 |
||||||||||
5 |
|
R |
|
|
|
|
|
N3 |
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||
|
|
|
||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
photoelimination |
|
|
|
R |
|
|
|
|
ONO |
|
|
|
|
* |
R |
|
|
|
|
O |
|
+ NO |
|
|||||||||||||||||
6 |
|
|
|
|
|
|
|
|
|
|
|
Photofragmentation/ |
6.4.2 |
|||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
* |
|
|
|
N O |
|
photoelimination |
|
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
7 |
|
|
|
|
|
|
|
O |
|
|
|
|
|
R |
|
|
Photorearrangement |
6.4.2 |
||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
R |
N |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R' |
|
|
|
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R' |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
* |
|
|
|
|
|
|
|
+ NO2 |
|
|
|||||||||||||||||
8 |
|
R |
|
|
NO2 |
|
|
|
R |
|
|
|
|
|
|
Photofragmentation |
6.4.3 |
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
9 |
|
R |
|
|
NO2 |
|
|
* + [H] |
R |
|
NO2H |
|
Photoreduction |
6.4.3 |
||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
||||||||||||||||||||||||||||||||
10 |
R3N |
|
|
|
|
|
|
+ [A] + hν |
R3N |
+ |
A |
Electron transfer |
6.4.4 |
|||||||||||||||||||||||||
11 |
Ar(CN)x + [D ] + hν |
Ar(CN)x + D |
Electron transfer |
6.4.4 |
||||||||||||||||||||||||||||||||||
a[H] ¼ hydrogen atom donor; [A] ¼ electron acceptor; [D] ¼ electron donor.
Nitrogen Compounds |
343 |
fragment via a homolytic cleavage to release a nitrogen molecule (entry 2) if the corresponding bond dissociation energies are sufficiently low. Fragmentation is the most common photoreaction of other chromophores, such as 3H-diazirines, diazo compounds (entry 3), diazonium salts (entry 4), azides (entry 5) and some heteroaromatic compounds (Section 6.4.2). Some of these processes are particularly efficient because thermodynamically stable and photochemically inert molecules, such as N2 or NO, are liberated. The primary photochemical process of organic nitrites is also homolytic fission, in this case of the N O bond (entry 6). The subsequent isomerization to the corresponding oximes is called the Barton reaction. The p,p excited nitrones or heterocyclic N-oxides typically undergo E–Z isomerization and competing rearrangement to form oxazirine derivatives (entry 7).
Photofragmentation is the principal primary process of excited aliphatic nitro compounds subsequently producing NO2 and the corresponding radicals (entry 8), which may recombine to form nitrites. The excited nitroalkanes or nitroarenes can also be photoreduced in the presence of a hydrogen-atom donor (entry 9).
Amines are generally good electron donors. They readily undergo photoinduced electron transfer (PET) processes, in which amine donates an electron to the reaction partner either in its ground or excited electronic state (entry 10). In contrast, electrondeficient, nitrogen-containing molecules, such as aromatic nitriles, may serve as electron acceptors (entry 11). Many organic metal complexes can also be involved in photochemically initiated redox reactions (Section 6.4.4).
6.4.1Azo Compounds, Imines and Oximes: E–Z Photoisomerization
|
|
|
|
hν |
|
|
|
|
|
|
|
hν |
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
R'' hν or ∆ |
|
|
||||
|
|
|
hν or ∆ |
|
|
|
|
|
|
R'' |
||||||||
|
|
|
|
|
||||||||||||||
Recommended review articles.1061–1070
Selected theoretical and computational photochemistry references.16,534,1071–1079
Like alkenes (Section 6.1.1), chromophores containing the N¼N (azo compounds) or C¼N (imines, oximes, etc.) bonds can undergo E–Z (or trans–cis) photoisomerization (Scheme 6.156) and the resulting isomer concentration ratio in the photostationary state (PSS) reflects the absorption properties of the isomers and isomerization quantum yields (see Scheme 6.1 in Section 6.1.1). Since conventional (dark) synthesis generally provides
access to more stable E-isomers, photochemistry is an exceptional tool for preparing sterically hindered Z-isomers.1061,1062 The photoisomerization reaction can be induced by a
direct irradiation or by sensitization and it often competes with other phototransformations, such as photofragmentation or photorearrangement (Section 6.4.2).
Unlike alkenes, the unsaturated azo group in N¼N or C¼N derivatives possess an in-
plane lone electron pair in an n-orbital. As a result, both the n,p and p,p excited states can be involved in two limiting mechanisms of photoisomerization.1062,1080,1081 The
first mechanism is a 180 rotation (twist) about the former double bond in conjunction with a reduced bond order, similar to that observed in alkenes (Scheme 6.157). In the second case, an in-plane inversion proceeds due to rehybridization of one nitrogen without
344 |
Chemistry of Excited Molecules |
||||||||||
|
|
|
|
|
|
E-Z |
|
|
|
||
|
|
|
|
isomerization |
|
|
|
||||
|
R1 |
|
hν (ΦE-Z) R1 R2 |
||||||||
|
N |
|
N |
|
|
|
|
|
N |
|
N |
|
|
|
|
|
|
|
|
||||
|
|
|
hν (ΦZ-E) |
|
|||||||
|
|
|
R2 |
|
|
||||||
|
|
|
|
|
|||||||
|
|
|
|
|
|
|
|||||
|
E-isomer |
|
|
|
|
|
Z-isomer |
||||
Scheme 6.156
any significant change in the bond order. The isomerization pathway is dependent on the excitation type. Quantum chemical calculations and also time-resolved experiments suggest
that a rotational E–Z interconversion in imines and azo compounds occurs from their 1n,p and 3n,p states. This remains a subject of intense interest.1062,1072,1082
C N H
H
out-of-plane
inversion
C N
H
 C N
C N H
H 
 in-plane inversion
in-plane inversion
Scheme 6.157
Azo Compounds
Even in the absence of p-delocalization, azoalkanes absorb in the near-UV region, although the corresponding molar absorption coefficients (n,p ) are small. In the absence of competing photoreactions, some rare sterically hindered Z-isomers can be prepared. For
example, (Z)-1,10-azodinorbornane (352) was prepared in toluene at 0 C from its E-precursor (Scheme 158).1083
hν |
N N |
|
|
N N |
|
hν |
|
|
352 |
Scheme 6.158 |
|
Azoalkanes may readily undergo photoelimination of N2 (Section 6.4.2) when stable radical intermediates are produced. For example, whereas irradiation in the n,p absorption region (>400 nm) of the cyclic 1,4-dihydronaphthodiazepine 353 induces E–Z isomerization, nitrogen eliminates to yield acenaphthene exclusively upon p,p excitation of the naphthalene moiety at 313 nm (Scheme 6.159).1084
The photophysical and photochemical properties of cyclic azo compounds, where Z ! E isomerization is suppressed by geometric constraints, are very sensitive to seemingly minor structural changes (Table 6.16). In contrast to 2,3-diazabicyclo[2.2.1]
Nitrogen Compounds |
345 |
||
N N |
|
N |
|
|
|
|
|
405 nm |
N |
|
|
436 nm |
|
|
|
or |
∆ |
|
|
353 |
|
|
|
313 nm |
- N2 |
|
|
Scheme 6.159
hept-2-ene (DBH), 2,3-diazabicyclo[2.2.2]oct-2-ene (DBO) possesses a remarkably long singlet lifetime, a high fluorescence quantum yield and a low quantum yield of denitrogenation, which has been exploited, for example, to measure the kinetics of conformational changes in biopolymers (Special Topic 2.3). The tricyclic diazabicycloheptene (fused DBH) is exceptional in exhibiting efficient intersystem crossing, a relatively long triplet lifetime and strong phosphorescence.1085
Table 6.16 Photophysical properties of bicyclic azoalkanes1085 |
|
|
|||
Property |
|
|
|
|
N |
|
|
|
|
|
|
|
N |
|
N |
N |
|
|
N |
N |
|
|
|
|
|
|
|||
|
(DBH) |
(DBO) |
(fused DBH) |
||
|
|
|
|
||
|
|
|
|
|
|
1t/ns |
0.15 |
690a |
|
2.4 |
|
3t/ns |
7b |
25c |
|
630 |
|
FISC |
0 |
0 |
d |
0.5 |
|
Ff |
<0.001 |
0.37 |
|
0.02 |
|
F N2 |
1.0 |
0.02d |
0.59 |
||
|
|
|
|
|
|
Fph (77 K) |
0.00 |
0.00 |
|
0.052 |
|
ES/(kJ mol 1) |
354b |
318b |
|
327 |
|
ET/(kJ mol 1) |
260d |
222d |
|
261 |
|
aRef. 1059. bRef. 1086. cRef. 1087. dRef. 1088.
Differences in absorption of (E)- and (Z)-azobenzene (Figure 6.9) enable us to select the irradiation wavelength to obtain the Z-isomer preferentially;559,1089,1090 however,
the photostationary concentration ratio of the isomers is affected by competing thermal Z ! E isomerization (Ea 100 kJ mol 1, compared with Ea 176 kJ mol 1 for
stilbene). Many (Z)-azobenzene derivatives can be isolated successfully at lower temperatures by chromatographic separation.1061,1062,1091 Since elimination of nitrogen to
give unstable aryl radicals from azobenzene is negligible, E–Z isomerization is usually the principal photochemical process observed, which is valuable for many applications.