

5.2. Хромосомные болезни с нарушениями психики
Хромосомные болезни обусловлены мутацией наследственного материала в половых клетках одного или обоих родителей на хромосомном или геномном уровне. Как правило, не наследуются, не столь разнообразны фенотипически (как генные), характеризуются тяжёлыми нарушениями психики в сочетании с рядом дефектов соматического развития.
Встречаются хромосомные болезни с частотой 1 : 250 (или 0.4% новорождённых, у эмбрионов до 4%, и 20% - среди спонтанных абортов). Лишь менее 10% хромосомных аномалий сбалансированы и особи жизнеспособны.
Олигофрения наблюдается при большинстве хромосомных синдромах: Дауна, Патау, Эдвардса, «кошачьего крика», Вольфа, Клайнфельтера, полисомии по Х-хромосоме, Тернера-Шерещевского и других.
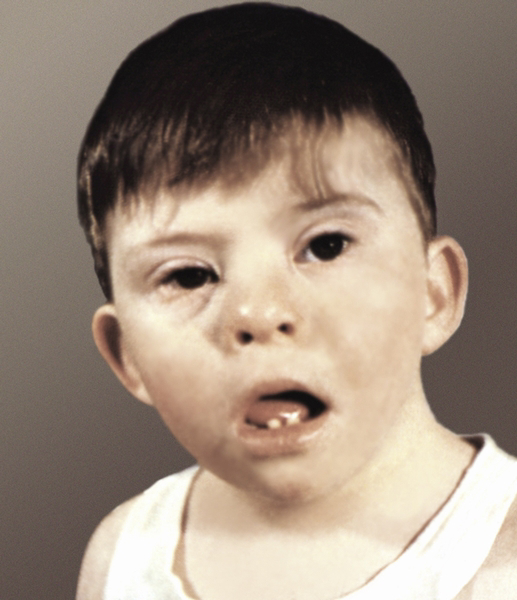
Болезнь Дауна, монголизм, синдром Дауна или трисомия по 21-ой паре хромосом обусловлена аномалией хромосомного набора; впервые была описана английским врачом Джоном Лангдоуном Дауном в 1866 году. Одна из наиболее распространенных хромосомных болезней, встречается в среднем с частотой 1 : 700, оба пола поражаются с одинаковой частотой, как правило, не наследуется, относится к врождённой патологии развития.
Многочисленными исследователями показано, что болезнь Дауна может быть в 91% случаев обусловлена трисомией 21-ой хромосомы, в 4% случаев транслокацией 21-ой хромосомы на 13-ую или 22-ую хромосому и в 2% случаев мозаицизмом, когда одна часть клеток имеет нормальный кариотип из 46 хромосом, а другая часть – 47.
Клиническая картина болезни Дауна, обусловленная трисомией или транслокацией, не различается. При синдроме Дауна, вызванном мозаицизмом, симптомы заболевания выражены менее резко.
Причины рождения детей с болезнью Дауна окончательно не выяснены. Предполагают, что причиной её могут быть перенесённые матерью перед зачатием инфекционные заболевания (гепатит, токсоплазмоз, корь, краснуха и другие)
Наблюдается сильная зависимость частоты синдрома от возраста матери. У потомства молодых (19 – 35 лет) матерей она низкая – 1 : 1 000, в 19 лет – 1 : 1 640, у малолетних матерей (до 19 лет) – она выше (1 : 500), у тридцатилетних - риск рождения ребенка с синдромом Дауна соответствует среднему популяционному риску (1 : 700), но уже к 40 годам, как показывает статистика, вероятность заболевания у ребенка достигает 1 : 200 – 1 : 100, в возрасте 40 – 41 год – 1 : 84, а затем еще более повышается и к 45 годам составляет 1 : 31.
Данные по зависимости рождений больных детей от возраста отца несколько противоречивы. В последние годы благодаря флюоресцентному анализу показано, что не расхождение хромосом происходит и при сперматогенезе, то есть отец также может быть «виновником» в появлении трисомии и возникновении болезни Дауна (в 20 – 25% случаев).
Особенно большой риск рождения ребёнка с болезнью Дауна в семье, где отцу более 50 лет, а матери более 40.
Поскольку частота рождения детей с болезнью Дауна резко возрастает у женщин старше 35 – 40 лет, полагают, что дополнительная 21-я хромосома в большинстве случаев возникает в результате не расхождения хромосом во время созревания женской половой клетки.
При морфологическом исследовании нервной системы погибших больных характерны уменьшение размеров и массы головного мозга, недоразвитие лобных и других его долей, слабая дифференцировка борозд и извилин мозга. В ряде случаев встречаются аномалии развития головного мозга и крупных мозговых сосудов. Гистологически выявляется нарушение дифференцировки нервных клеток и недостаточная миелинизация нервных волокон головного и спинного мозга. Внутренние органы уменьшены в размерах. Наблюдается гипоплазия желез внутренней секреции, особенно щитовидной железы, коры надпочечников и половых желез. В печени — жировая вакуолизация, фиброз. Аорта узкая, стенки ее тонкие, крупные сосуды — меньшего диаметра. Часто отмечаются врожденные пороки сердца, желудочно-кишечного тракта и других органов.
Клиническая картина болезни хорошо изучена. В первые дни жизни ребёнка диагностика болезни наиболее трудная. В старшем возрасте она диагностируется легче. Такие дети имеют некоторые общие морфологические черты. Они больше похожи один на другого, чем на своих родителей.
Х
У маленьких детей резко выражена мышечная гипотония, из-за чего в лежачем положении живот приобретает форму «лягушечьего», отмечаются разболтанность суставов, «куриная» или «воронкообразная» грудь.
Патология скелета: низкий рост, укорочение дистальных отделов конечностей (короткая шея, стопы и кисти), полнота; кисти и стопы широкие и короткие (акромикрия); пальцы как бы обрублены, из-за гипоплазии средней фаланги мизинец укорочен и искривлен (клинодактилия), имеет одну сгибательную складку вместо нормальных двух; на ладони часто обнаруживают поперечную складку и высоко расположенный добавочный трирадиус (t’’) — точку, в которой сходятся папиллярные линии трех направлений; на стопах увеличен промежуток между I и II пальцами (valgus).
Н

Больные с рождения отстают в росте, поздно начинают держать голову, сидеть, ходить. Поздно и в неправильном порядке прорезываются зубы. Половое развитие резко задержано. Способность к деторождению установлена в единичных случаях. У многих больных имеются врожденные пороки сердца (до 50%); нередко отмечаются пороки развития различных отделов желудочно-кишечного тракта (атрезии, стенозы, мегаколон), иммунодефицитные состояния; у больных часто возникают злокачественные опухоли.
При синдроме Дауна продолжительность жизни резко снижена, обычно она составляет 20 — 25 лет. Однако развитие современной медицины позволяет им достигать и более зрелого возраста; описан больной с синдромом Дауна в возрасте 70 лет. По крайней мере 50% женщин с синдромом Дауна могут иметь детей. 35 – 50% детей, рождённых от матери с синдромом Дауна, рождаются с синдромом Дауна или другими хромосомными отклонениями.
В сыворотке крови обнаруживают увеличение концентрации иммуноглобулина G и снижение иммуноглобулина М. Характерны снижение сопротивляемости к инфекционным болезням, склонность к заболеванию лейкозом. По данным других авторов больные с синдромом Дауна реже имеют раковые опухоли. Видимо, 21-ая хромосома содержит ген – «глушитель опухолей», и наличие третьей копии гена обеспечивает дополнительную защиту против рака.
Изменения нервной системы доминируют в клинической картине болезни. У большинства больных окружность головы уменьшена, череп брахицефалической формы. С первых дней жизни ребенка выявляется мышечная гипотония; рефлекс Моро отсутствует. Отмечаются косоглазие, обычно сходящееся, слабость конвергенции, асимметрия лицевой иннервации, горизонтальный нистагм.
У части детей с болезнью Дауна обнаруживаются расстройства координации, которые проявляются при выполнении локомоторных проб, тонких движений, имеются дистрофические изменения костей, больные не уклюжи, не ловки, не координированы. С возрастом отмечается тенденция к нормализации мышечного тонуса, улучшается координация движений.
У всех больных снижена сопротивляемость к инфекциям, имеются вегетативно-эндокринные расстройства: сухость кожи, предрасположение к ожирению, дерматитам, красный стойкий дермографизм.
Психические расстройства характеризуются главным образом слабоумием по типу психического недоразвития – олигофрении, которая обнаруживается уже на первом году жизни. Отмечается диффузный характер слабоумия, при котором недоразвиты не только интеллект и мышление, но и другие психические функции (восприятие, внимание, память, речь, эмоционально-волевая сфера). Наряду с этим характерно преимущественное недоразвитие наиболее дифференцированных онтогенетически молодых функций — мышления и речи при относительной сохранности эволюционно более древних элементарных функций — эмоций и инстинктов.
Психическое недоразвитие при болезни Дауна в большинстве случаев на уровне имбециальности или дебильности, реже наблюдается идиотия. Суждения больных примитивны, абстрактное мышление им недоступно. Речь развивается поздно, словарный запас беден, произношение с дефектами, не могут писать и считать. Характерны замедление мышления, плохая переключаемость, больные легко теряются в непривычной обстановке. Внимание неустойчивое, легко отвлекаемое. Относительно хорошо развита механическая память, выражена подражательность. Эмоции сохранены, но мало дифференцированы, больные пассивны и несамостоятельны, отличаются повышенной послушностью и внушаемостью.
По особенностям темперамента чаще встречается вариант заболевания с преобладанием эретичности (возбудимости и раздражительности в сочетании с двигательным беспокойством), реже торпидности (вялости, пассивности и психомоторной замедленности).
В ряде случаев возможна адаптация их к жизни - они могут обслуживать себя, выполнять несложную работу, обучаемы несложным житейским навыкам, не требующим физической силы и инициативы. Дети с болезнью Дауна, как правило, очень ласковые и очень привязываются к своим близким.
Диагноз обычно несложен, в большинстве случаев устанавливается уже в родильном доме. При стертых клинических признаках необходимо цитогенетическое исследование.
При дерматоглифическом исследовании родителей детей с болезнью Дауна выявлены некоторые фенотипические особенности (осевой трирадиус Т, петли на гипотенаре, радиальные петли на IY и Y пальцах, аномальные ладонные борозды.
Специфических методов лечения пока не существует. Однако комплексная медикаментозная терапия в сочетании с лечебной физкультурой, массажем, педагогическим воздействием, занятиями с логопедом способствует улучшению состояния больных. Применяют различные методы стимуляции психического и физического развития — препараты ноотропного ряда, анаболические стероиды, витамины, тиреоидин, глутаминовую кислоту, липоцеребрин.
В развитых странах (США, Канада, Австралия и др.) накоплен богатый опыт умственного развития «даунов»: многие из них в состоянии считать и писать, имеют развитую речь с небольшими дефектами дикции. Богатые бездетные родители из этих стран спокойно берут «детей-даунов» из нашей страны на воспитание.
Прогноз для жизни относительно благоприятный; при тяжелых врожденных пороках сердца и желудочно-кишечного тракта и развитии лейкоза - неблагоприятный.
Профилактика включает МГК больного и родителей, особенно молодого возраста (возможность транслокации хромосом), для определения степени риска повторного рождения в этой семье больного ребенка.
Другие аутосомные трисомии встречаются гораздо реже. В 1960г. были описаны первые два случая заболевания по 13-ой и 18-ой хромосоме.
Синдром Эдвардса или трисомия по 18 хромосоме встречается с частотой 1 на 6 500 новорожденных, 50% детей доживает до 2-х месяцев, 1/3 – до 3 месяцев и только 1 – 2% - более года. Характерно, что, соотношение между девочки и мальчиками 3:1. Причина этого явления известна. Мальчики менее жизнеспособны, поэтому чаще погибают ещё в утробе матери.
При синдроме Эдвардса у детей наблюдаются большие изменения со стороны черепа и скелета. Такие дети обычно рождаются не доношенными (очень маленький рост), в асфиксии, с долихоцефалией, узким лбом, выступающим затылком, расщеплением нёба. Внешность больных с синдромом Эдвардса характерна: недоразвитая нижняя челюсть, рот маленький, узкие короткие глазные щели, маленькие глазные яблоки, деформированные низко расположенные уши, короткая шея, вытянутая голова с выдающимся затылком («птичий профиль»).
Отмечаются большие деформации пальцев рук, уплощение свода стопы, из-за чего стопа имеет форму качалки, пальцы ног укорочены, короткая грудина, дефектное развитие мускулатуры, физическая слабость.
Имеются пороки сердца, почек, желудочно-кишечного тракта, половых органов, демиелинизация больших полушарий и мозжечка, у мальчиков отмечены крипторхизм, а у девочек – гипертрофия клитора; глубокая дебильность. Кожа очень подвижна, из-за чего образует складки на шее и других частях тела
Мальчики погибают вскоре после рождения, девочки живут до года. Средний возраст матерей – 35 лет.
Синдром Патау или трисомия по 13 хромосоме встречается с частотой
1: 14 705. Впервые заболевание было описано в 1961г. К. Патау. Всего описано более 200 больных. Частота рождения мальчиков и девочек при синдроме Патау одинакова.
При синдроме Патау отмечаются значительные дефекты строения черепа и головного мозга: микроцефалия, недоразвитие переднего отдела мозга, атрофия обонятельных долей и зрительного тракта, низкий, скошенный лоб, узкие глаза, запавшая переносица, гипотелоризм, низко расположенные деформированные ушные раковины; часто встречается расщелина верхней губы («заячья губа») и нёба, ангиома лица (рисунок 34).
Б олее
чем у 70% больных наблюдаются пороки
органов зрения: микрофтальм или анофтальм,
глазные щели узкие или могут совсем
отсутствовать (при вскрытии глазных
щелей там обнаруживают колобому вместо
глазного яблока), катаракта.
олее
чем у 70% больных наблюдаются пороки
органов зрения: микрофтальм или анофтальм,
глазные щели узкие или могут совсем
отсутствовать (при вскрытии глазных
щелей там обнаруживают колобому вместо
глазного яблока), катаракта.
П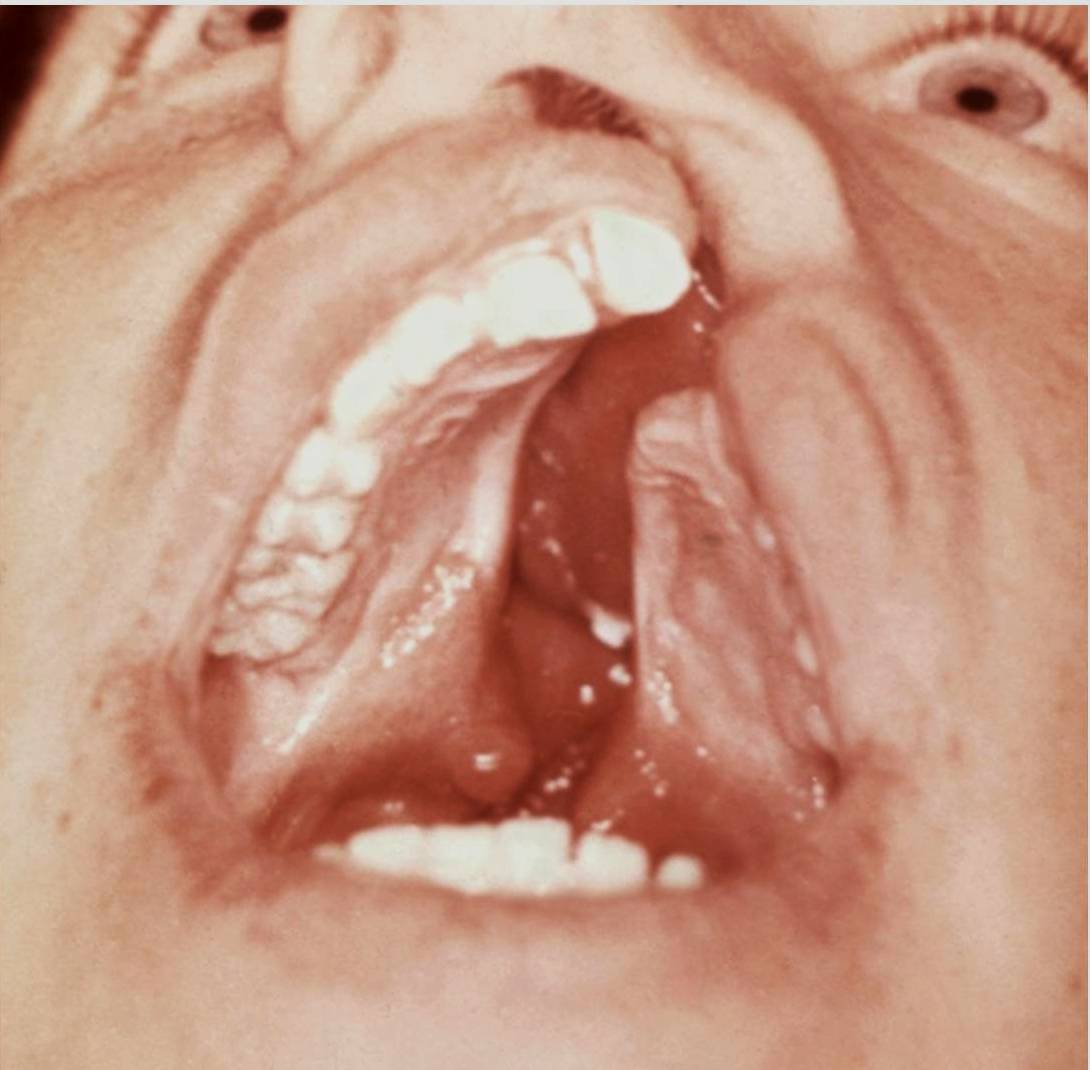 ри
синдроме Патау наблюдаются также
множественные пороки опорно-двигательного
аппарата и внутренних органов:
полидактилия, синдактилия, дефекты
сердечно-сосудистой системы (77%), аномалии
почек, желудочно-кишечного тракта,
половых органов, глубокая умственная
отсталость, судороги.
ри
синдроме Патау наблюдаются также
множественные пороки опорно-двигательного
аппарата и внутренних органов:
полидактилия, синдактилия, дефекты
сердечно-сосудистой системы (77%), аномалии
почек, желудочно-кишечного тракта,
половых органов, глубокая умственная
отсталость, судороги.
Дети редко доживают до 3 месяцев. Плод с признаками синдрома Патау обычно обнаруживается при спонтанных абортах. Средний возраст матерей – 32 года.
Трисомия по 22-ой хромосоме. В литературе описано около 30 случаев трисомии по 22-ой хромосоме. Эти дети – глубокие олигофрены. У них выражены микроцефалия, клювовидный нос, низко расположенные ушные раковины, расщепление нёба (рисунок 35), гипоспадия, гипотония мышц. Такие дети рождаются у родителей с нераспознанным мозаицизмом. Диагноз можно установить на основании исследования кариотипа.
Синдром полисемии по Х-хромосоме у женщин, синдром 47,ХХ+Х) или трисомия по Х-хромосоме или синдром 47,ХХ+Х встречается с частотой 0.1%. При кариотипе 47,ХХ+Х фенотипических изменений может и не быть, так как здесь две Х-хромосомы спирализованы и представляют собой половой хроматин. Пол – женский, девочки жизнеспособны, у 75% имеется лёгкая степень дебильности, велик риск (в 2 раза) заболеть психозом, особенно шизофренией. Нередка задержка физического развития, эпикант, высокое твёрдое нёбо, клинодактилия мизинцев, аменорея, преждевременный климакс, бесплодие.
Такие женщины могут иметь здоровое потомство, так как половина их гамет несёт нормальный набор хромосом.
В случае варианта этого синдрома – 48,ХХ+ХХ увеличивается глубина соматических аномалий. Описаны случаи с 4 и 5 хромосомами. Чем больше количество Х-хромосом, тем более выражен дефект умственного развития, а также изменения фенотипа и половой инфантилизм. Такие женщины высокого роста, со значительными изменениями скелета, искривлением позвоночника, депигментированными пятнами и т.д. Чем больше Х-хромосом в кариотипе, тем меньше гребневый счёт, так как у них преобладают дуговые узоры на кончиках пальцев. При кариотипе 48,ХХ+ХХ дети мало жизнеспособны и обычно рано умирают.
Сравнивая между собой все случаи трисомий учёные установили, что чем крупнее (больше размер) хромосома и, следовательно, меньше её порядковый номер, тем тяжелее последствия такой структурной перестройки для больного ребёнка.
Синдром «кошачьего крика» – хромосомное заболевание, которое сопровождается врождённой патологией голосовых связок, заметной при рождении и нивелирующейся в период полового созревания. Причиной синдрома «кошачьего крика», впервые описанного в 1963г., является делеция короткого плеча 5-й хромосомы (кариотип 46,ХХ,5р~ или 46,XY,5p~). Частота его среди новорожденных составляет 1 : 45 000, причем девочки рождаются немного чаше.
Наиболее характерным признаком заболевания является необычный плач, особый, жалобный тембр голоса, похожий на требовательное кошачье мяуканье или крик. Причиной его являются патологические изменения гортани.
У этих детей наблюдается также микроцефалия, микрогения, лунообразное лицо с уменьшенной верхней челюстью и складками в уголках глаз, высокое нёбо, плоская спинка носа, низко расположенные деформированные уши, гипертелоризм, антимонголоидное направление глазных щелей, эпикант, поперечная складка на ладони.
Кроме того, встречаются врождённые пороки сердца, пороки внутренних органов, мышечная гипотония, патология костно-мышечной системы, деформация стоп, включая сращение соседних пальцев (синдактилию). Девочки болеют чаще мальчиков.
Выраженность клинической картины меняется с возрастом. «Кошачий крик», мышечная гипотония, лунообразность лица с возрастом исчезают, а микроцефалия выявляется более отчётливо, прогрессирует психомоторное недоразвитие, косоглазие.
Все больные характеризуются тяжелой степенью умственной отсталости. Хотя продолжительность жизни больных с синдромом «кошачьего крика» выше, чем у страдаюших аутосомными трисомиями, тем не менее, большинство из них умирает в раннем возрасте. Вместе с тем описаны больные старше 50 лет. У выживших больных с синдромом «кошачьего крика» дефект голоса с возрастом (к моменту начала полового созревания) исчезает, а умственная отсталость остаётся (вплоть до глубокой дебильности).
Синдром делеции длинного плеча 18-ой хромосомы – типичный пример хромосомной аберрации. Клинические проявления при делеции длинного плеча 18-ой хромосомы разнообразны и включают изменения со стороны нервной системы, сердца, костей, глаз и наружных половых органов.
В общем виде они могут быть представлены следующим образом: 1) со стороны нервной системы – умственная отсталость (около 100%); 2) костно-мышечная система – низкий рост (80%), микроцефалия (100%), дисплазия средней части черепа (плоская переносица, маленький тупой нос, ложный прогнатизм, гипертелоризм, эпикант, высокое нёбо, «карпий рот», неправильный рост зубов, широкая грудная клетка, брахицефалия, веретенообразные пальцы, косолапость и плоскостопие; 3) уши – низкое расположение, изменение формы ушной раковины (скрученный завиток, выступающие противозавиток и противокозелок), отсутствие или сужение слухового прохода; 4) глаза – спонтанный нистагм, косоглазие, аномалии глазного дна, глаукома, дисплазия передней камеры глаза, микророговица, колобома; 5) наружные половые органы – недоразвитие или отсутствие малых половых губ у девочек, гипоплазия полового члена и крипторхизм у мальчиков; 6) сердце – врождённый порок.
Клинический полиморфизм, выявляющийся при этом, многие авторы объясняют поражением различных участков 18-ой хромосомы. Наличие же общих признаков позволяет объединить их под общим названием – синдром делеции длинного плеча 18-ой хромосомы.
Синдром Клайнфельтера (синдром 47,ХY+X, 48, XY+XX) впервые был описан в 1942г. H. Klinefelter, E. Reifenstein и F. Albriglit. В 1956г. Бриге и Барр выявили в кариотипе таких больных лишнюю Х-хромосому. Синдром Клайнфельтера встречается с частотой 1 : 500, пол – мужской. Мальчики жизнеспособны. Заболевание диагностируется, как правило, в период полового созревания, когда отмечаются признаки евнухоидизма (рисунок 36). Выделяются два типа телосложения: для одних больных характерен высокий рост с астеническими чертами телосложения, для других – евнухоидные пропорции и гинекомастия. У всех больных не доразвиты тестикулы при удовлетворительном развитии полового члена. Нет растительности на лице, выражена гинекомастия, отмечается отложение жира на бёдрах, как у женщин, рост волос на лобке по женскому типу, высокий голос. Больные высокого роста за счёт удлинения ног при относительно коротком туловище, руки у них длинные, кисти достают до колен. У больных обычно отмечается умственная отсталость разной степени выраженности, но встречаются и лица с нормальным интеллектом. Такие больные обычно бесплодны.
К внешним порокам развития добавляется
задержка умственного развития (дебильность
средней степени), малоинициативность,
отсутствие тяги к творчеству, лёгкая
внушаемость, эмоциональная неустойчивость,
аффектность поведения, иногда
агрессивность.
внешним порокам развития добавляется
задержка умственного развития (дебильность
средней степени), малоинициативность,
отсутствие тяги к творчеству, лёгкая
внушаемость, эмоциональная неустойчивость,
аффектность поведения, иногда
агрессивность.
Наличие таких признаков, особенно лёгкой внушаемости, на практике приводит к тому, что такие больные оказываются очень часто втянутыми в различные религиозные секты, бандформирования, совершают террористические акты, как комикадзе (с поясом шахидов), поэтому больные с синдромом Клайнфельтера представляют большую социальную опасность.
Вариант синдрома Клайнфельтера 48 (ХY+ХХ, тетрасомия) редко встречается среди умственно отсталых детей. Поражение интеллекта более выражено. Мальчики имеют очень высокий рост (+15см), евнухоидное сложение, гинекомастию, микроорхидизм, варикоз, среднюю степень дебильности и очень агрессивны.
Вариант синдрома Клайнфельтера 49 (ХY+ХХХ) встречается чаще, чем 48. при этом добавляются скелетные аномалии. Чем больше Х-хромосом в кариотипе больных, тем больше выражены фенотипические признаки заболевания и степень дебильности.
Этиология болезни Клайнфельтера неясна. Такие дети чаще рождаются у пожилых матерей, хотя кариотип их нормален. Нерасхождение Х-хромосом, по-видимому, происходит на ранних стадиях дробления зиготы.