

4.3.2. Хромосомные заболевания
В соматических клетках человеческого организма имеется диплоидный набор хромосом – 23 пары (46 хромосом), а в половых клетках (гаметах) – гаплоидный (одинарный) набор хромосом (23 хромосомы). У мужчин и женщин 22 пары диплоидного набора соматических клеток, одинаковы по форме и величине и называются аутосомами, 23-я пара – половые хромосомы (гоносомы, гетеросомы) представлена у женщин двумя Х-хромосомами, ау мужчин – одной Х- и другой Y-хромосомой. Все яйцеклетки несут одинаковый набор хромосом (моно-гаметный пол). В одну половину образующихся спермиев идёт 22 аутосомы и одна Х-хромосома, а в другую половину – 22 аутосомы и одна половая Y-хромосома (гетерогаметный пол).
Х
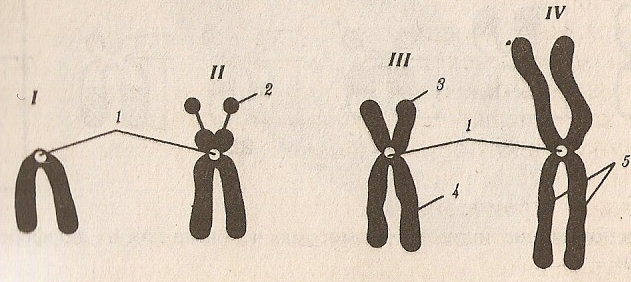
1-ая группа (А), 1, 2 и 3-я пары – крупные метацентрические хромосомы, в которых центромера находится по середине (на рисунке 19 – тип IY).
2-ая группа (В), 4 и 5-ая пары – крупные субметацентрические хромосомы, в которых центромера смещена к одному концу (на рисунке 19 - тип III).
3-я группа (С), 6 – 12-ая пары хромосом и половая Х-хромосома – средние субметацентрические хромосомы (на рисунке 19 - тип III).
4-ая группа (D), 13, 14 и 15-ая пары – акроцентрические хромосомы, в которых центромера находится на конце хромосомы (на рисунке 19 - тип II)
5-ая группа (Е), 16, 17 и 18-ая пары – малые субметацентрические хромосомы.
6-ая группа (F), 19 и 20-ая пары – малые метацентрические хромосомы.
7-ая группа (G), 21 22-ая пары и половая Y-хромосома – малые акроцентрические хромосомы.
Все 7 групп хромосом образуют полный набор хромосом или кариотип человека. Исследование набора хромосом с помощью микроскопа называется кариотипированием.
Таким образом, женский кариотип обозначается, как 46,ХХ, а мужской – 46,ХY (рисунок 20).
Установлено, что пол будущего ребёнка в основном зависит от Y-хромосомы, в которой расположен ген, определяющий дифференцировку половых желёз по мужскому типу и отвечающий за синтез тестостерона.
Однако этого не всегда достаточно для развития мужского организма. Для этого необходим также белок-рецептор, обеспечивающий проникновение гормона в клетки тканей-мишеней. За синтез такого белка отвечает особый ген, расположенный в аутосоме. Его мутация, нарушающая образование нормального белка-рецептора, делает ткани-мишени невосприимчивыми к тестостерону.
Не использовав такую возможность на определённом этапе онтогенеза, организм осуществляет развитие по женскому типу. В результате появляется особь с кариотипом XY, но внешне более сходная с женщиной. Такие субъекты не способны иметь потомство, так как их семенники недоразвиты, а их выводные протоки часто формируются по женскому типу (недоразвитая матка, влагалище). Вторичные половые признаки также характерны для женского пола. Описанная картина известна как тестикулярная феминизация или синдром Морриса.
Другим доказательством утверждения, что только Y-хромосома является половой, послужили результаты исследований с использованием современных технологий. Так, при помощи электронного микроскопа и микропипетки удалось изъять Y-хромосому на самых ранних этапах эмбрионального развития из зиготы. В результате пол (мужской) организма не изменился. Замена Х-хромосомы на Y-хромосому приводил к смене пола. Аналогичные манипуляции с Х-хромосомой к смене пола не приводили. Поэтому в последнее время принято считать, что у человека одна, а не две, как считали раньше, половая хромосома.
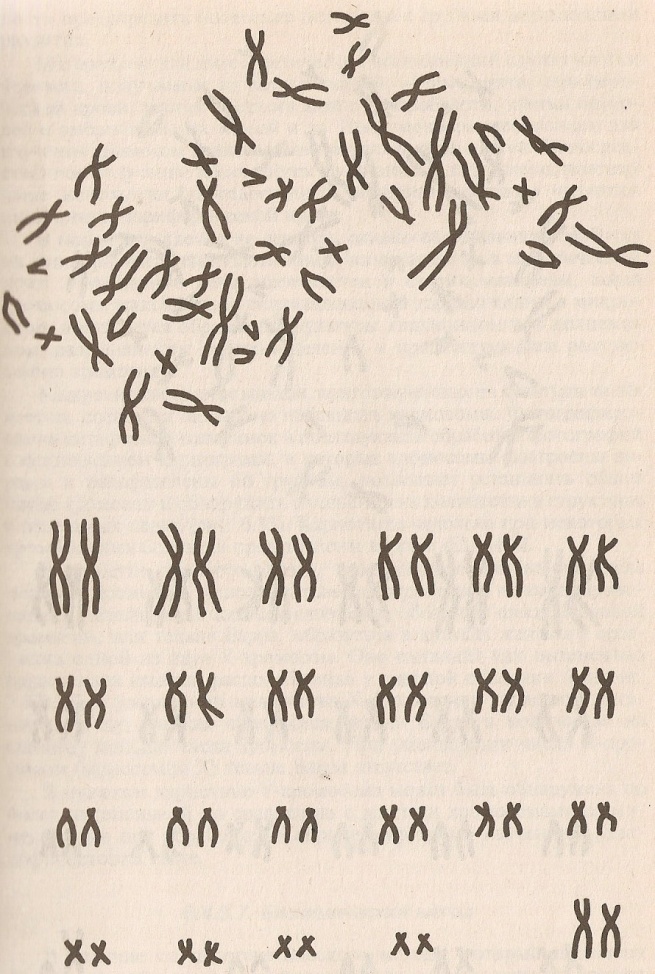
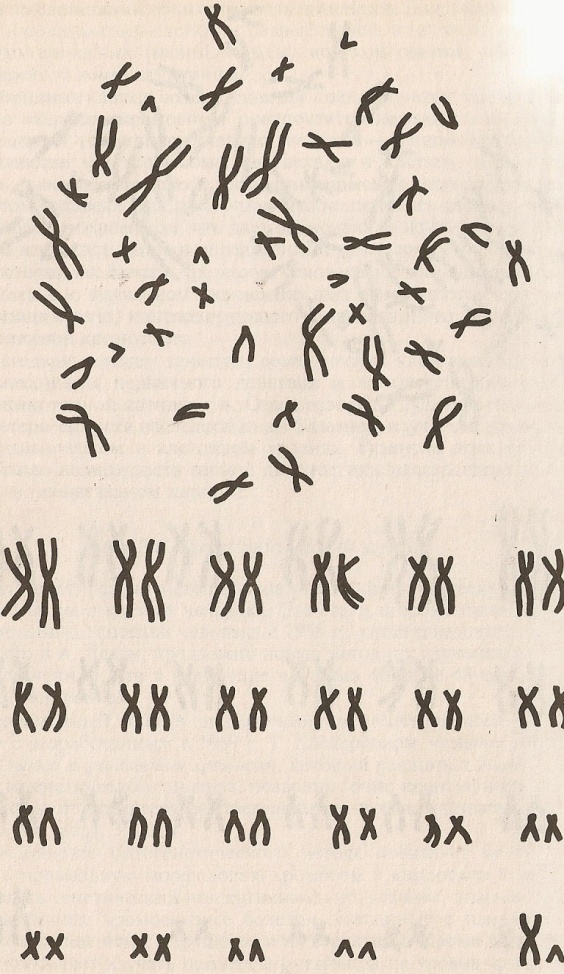
Рисунок 20. Женский (слева) и мужской (справа) набор хромосом.
Истинный гермафродитизм – чрезвычайно редкое заболевание (в мировой литературе описано всего около 150 случаев). Ложный гермафродитизм включает все формы тестикулярной и экстрагенитальной (надпочечниковой, медикаментозной и др.) патологии полового развития.
Гермафродитизм – типичная врожденная патология. При количественных или качественных изменениях в хромосомном наборе нарушается формирование гонад (дисгенезия гонад): они не образуются вообще (агенезия гонад) или содержат герминативные структуры обоих полов, являясь неполноценными анатомически и функционально.
Герминативные структуры гонад мужского (семенные канальцы) и женского (фолликулы) пола могут быть сгруппированы в одной гонаде (ovotestis). У подавляющего большинства гермафродитов зрелого возраста функционально превалирует овариальная часть гонады, о чем свидетельствует появление менструаций. Вторая гонада может быть однополой (яичник или яичко) или двуполой. Возможна также комбинация яичника с одной и яичка с другой стороны.
Истинный гермафродитизм, по данным литературы, характеризуется женским набором половых хромосом (46,ХХ), иногда при гермафродитизме встречаются различные формы мозаики; мужской набор хромосом (46,XY) при истинном гермафродитизме отмечают редко. У истинных гермафродитов обычно имеются матка, маточные трубы, своды влагалища. При наличии яичка с одной стороны матка формируется однорогой – со стороны оставшегося яичника.
Вторичные половые признаки у таких субъектов обычно имеют элементы обоих полов: низкий тембр голоса, смешанный тип фигуры, в той или иной мере развитые молочные железы и оволосение по мужскому типу. Зачатия у достоверных гермафродитов в литературе не описано, хотя наличие матки и овуляции свидетельствует о такой возможности.
Одним из важных лечебных мероприятий является хирургическое изменение пола. Перемена пола у детей 3 – 4 лет не вызывает психологических трудностей. В возрасте 4 – 10 лет такая перемена переносится тяжело, т.к. обычно в этом возрасте твердо устанавливается сознание половой принадлежности, а понятия пола в смысле половой функции еще не существует. Начиная с пубертатного периода, дети осознают свою интерсексуальность и становятся еще более ранимыми. Нередко они сами настаивают на определении пола.
В юношеском возрасте (16 – 17 лет) эта проблема осложняется в связи с появлением тщательно скрываемых и подавляемых половых влечений, обычно гетеросексуальных по отношению к тому полу, в принадлежности к которому воспитывался больной. В целом у больных в возрасте 16 – 20 лет адаптация к перемене женского пола на мужской психологически происходит легче, чем при перемене мужского на женский, т. к. особенности мужского поведения приобретаются легче. Смена пола в более старшем возрасте затруднительна по социальным причинам (приобретение определенной специальности, общественного положения и т.п.)
При выборе пола главным образом учитывают функциональные особенности наружных половых органов и функциональное превалирование женской или мужской части двуполой гонады. Элементы гонады противоположной избранному полу, по возможности удаляют. Производят хирургическую коррекцию наружных половых органов.
Выяснилось, что у человека в утробе матери на ранних этапах эмбриогенеза под влиянием сильных внешних факторов могут произойти разнообразные хромосомные мутации, перестройки или аберрации, приводящие к изменению количества или структуры хромосом. Все хромосомные аберрации приводят к серьёзным последствиям, в частности к нарушению развития строения и функций организма. По этой причине имеется целая группа врождённых заболеваний, названных хромосомными болезнями или синдромами, аберрациями. Доказано, что некоторые хромосомные перестройки, произошедшие в половых клетках до оплодотворения, могут передаваться по наследству, поэтому считаются наследственными.
Во внутриутробном развитии человека различают (условно) два периода: зародышевый (эмбриональный) и плодовый (фетальный). Эмбриональный период продолжается от момента оплодотворения яйцеклетки сперматозоидом и до конца второго месяца беременности. В этот период образуются зачатки всех важнейших органов и систем (нервная, кроветворная, сердечно-сосудистая, пищеварительная, выделительная, эндокринная и др.); происходит формирование туловища, головы, лица, зачатков конечностей. Зародыш приобретает черты, характерные для человека. Процессы развития в этот период весьма интенсивны, приспособительные механизмы ещё не развиты, поэтому зародыш очень чувствителен к действию повреждающих факторов.
Фетальный период начинается с конца второго месяца и продолжается до рождения плода. В этот период происходят быстрый рост плода, дифференцировка тканей, развитие органов и систем.
Известно, что около 170 из 1 000 эмбрионов и плодов погибают до рождения, из них около 40 % – вследствие несовместимых с жизнью хромосомных дефектов. Примерно 20 % нежизнеспособных выкидышей и абортусов (абортированные плоды) имеют хромосомные перестройки. Тем не менее, значительная часть мутантов (носителей хромосомной аномалии) минует действие внутриутробного отбора. Но некоторые из них погибают в раннем детстве. Больные с аномалиями половых хромосом из-за нарушения полового развития, как правило, не оставляют потомства. Частота хромосомных болезней в Томской области составляет более 2.5 %.
Хромосомные болезни, строго говоря, не являются наследственными в том смысле, что они в большинстве случаев не передаются по наследству. Кариотип родителей этих больных обычно нормальный, а аномальные хромосомные изменения обычно происходят в гаметах, из которых развивается больной организм. Но, так как именно в хромосомах находится наследственная информация, то хромосомные болезни по механизму своего появления следует трактовать как наследственные, поскольку в их основе лежит нарушение аппарата наследственности.
Хромосомные заболевания, как правило, не столь разнообразны фенотипически, как генные, характеризуются тяжелыми нарушениями психики в сочетании с рядом дефектов соматического развития.
Общими проявлениями хромосомных аномалий являются: черепно-лицевые дисморфии, задержка физического и психомоторного развития, умственная отсталость, костно-мышечные аномалии, пороки сердечно-сосудистой, мочеполовой, нервной и других систем, отклонение в гормональном, биохимическом и иммунологическом статусе. К настоящему времени известно более 100 хромосомных синдромов.
Лишь 10 % хромосомных аномалий сбалансированы и дети выживают. Степень поражения органов при хромосомных болезнях зависит от многих факторов – типа хромосомной аномалии, недостающего или избыточного генного материала, генотипа организма, условий среды, в котором развивается организм. Этиологическое лечение хромосомных болезней в настоящее время не разработано.
Из причин, вызывающих хромосомные болезни, наиболее значимые четыре первых.
Первая причина – возраст матери или состояние ее здоровья на момент зачатия (с возрастом обычно ухудшается). Так, средний возраст матерей, у которых родился первый ребёнок, по данным родильных домов в США составляет 28 лет, средний возраст матерей, рожающих синдром Дауна – 33 года и два хромосомных дефекта одновременно – 42 года.
Подсчитано, что риск рождения ребёнка с синдромом Дауна составляет в 16 лет – 1 : 500, в 19 лет – 1 : 1 600, в 25 лет – 1 : 1 250, в 30 лет – 1 : 952, в 35 лет – 1 : 378, в 40 лет – 1 : 106, в 45 лет – 1 : 30 и в 49 лет – 1 : 11.
Научные исследования также показали, что к группе риска относятся девушки, забеременевшие до 16 лет. Это связано как с уменьшением гормонального контроля оогенеза у пожилых женщин, так и с недостаточностью гормонального контроля у девушек подросткового возраста. Риск рождения ребёнка с синдромом Дауна также увеличивается при пожилом отце (60 – 75 лет).
В связи с этими статистическим данными американские учёные сделали вывод о зависимости частоты хромосомных аномалий у детей от возраста матери.
В нашей стране ситуация с цифрами отличается только сдвигом возраста матери в среднем на 7 лет в меньшую сторону и значительно более высокой частотой рождения детей с хромосомными аномалиями.
Причина такой существенной разницы лежит в том, что в нашей стране государство не борется с малолетним материнством, просто игнорирует это явление. Поэтому для Росси возрастной аспект здоровья женщины требуется рассматривать двояко. Первая группа проблем возникает у малолетних матерей, то есть у девушек до 21 лет. Вторая группа проблем связана со «старородящими» женщинами, то есть с женщинами, рожающими первого ребёнка после 30 лет.
Более 40 лет назад в развитых странах мира (в первую очередь США, Канада, Австралия, страны европейского экономического сообщества) на государственном уровне были разработаны и успешно применены методики профилактики малолетнего материнства, которое приводит к появлению большого количества проблем со здоровьем, развитием и воспитанием детей.
Детям с малых лет на различных уровнях, используя разнообразные формы пропаганды (в первую очередь телевидение), внушали, что необходимо планирование семьи, планирование рождения ребёнка, что секс и любовь (брак) – разные понятия. Ключевой момент психологического воздействия на молодежь (особенное во времена сексуальной революции на Западе) состоит в том, что молодёжи внушается, что иметь детей раньше 24 лет не имеет смысла, по крайней мере, по трём важным причинам:
1. До 24 лет человека нельзя считать полностью взрослым, так как он «не созрел» физиологически. К этому времени ещё только заканчивается половое созревание человека и, следовательно, мужчина и женщина не достигли своего физического (полового) совершенства, поэтому нет гарантии полноценного потомства. По этой причине при выборе в качестве суррогатной матери (из 3 вариантов: 16, 24 и 40 лет) для вынашивания своей оплодотворённой яйцеклетки все женщины, имеющие проблемы с вынашиванием плода, останавливаются на 24-летней женщине.
Имеется аналогичный пример, связанный с половым здоровьем мужчины. В случае выбора донора спермы все женщины выберут для оплодотворения своей яйцеклетки сперматозоиды здорового 24-летнего мужчины.
2. К 24 годам человек, как правило, получает образование, работу, квартиру и может содержать семью, создать благоприятные условия для воспитания детей. То есть до 24 лет человек «не созрел» экономически.
3. До 24 лет человек не может иметь полноценную семью, так как «не созрел» психологически. То есть он ещё ребёнок, который должен доиграть все свои игры (в зависимости от возраста), посетить все свои дискотеки и т.д.
Внимательно вглядитесь в американские сериалы (типа «Санта-Барбара»), американские фильмы для молодёжи и вы увидите, море секса. Но нигде не встретите малолетнюю мать в качестве героини фильма для молодёжи. В крайнем случае, речь будет вестись о том, что она забыла о доступной контрацепции и теперь срочно надо искать деньги на проведение дорогостоящего и опасного для здоровья аборта.
В сериалах для взрослых мы видим планирование рождение ребёнка. Ребёнок появляется только в семье, имеющей все возможности (квартиру, зарплату) для полноценного воспитания. Беременная мать все 9 месяцев занимается в спортзале, делая специальную гимнастику для беременных, или бегает кроссы (в США), плавает в бассейне (в Японии). Такая пропаганда медицинских знаний принесла свои плоды. В развитых странах Запада малолетнее материнство не носит характера эпидемии, что сказывается на среднем показателе возраста женщин при рождении первого ребёнка (в США – 29 лет, в России – 21 год)
Вторая причина хромосомных болезней – ионизирующая радиация. Третья причина – вирусные инфекции (корь, краснуха, ветряная оспа, герпес zoster, желтая лихорадка, вирусный гепатит, токсоплазмоз и вплоть до кариеса зубов и гриппа).
Четвертая причина – болезни эндокринной системы, особенно щитовидной железы (тиреотоксикоз), надпочечников (выброс кортикостероидов при стрессе) и половых желез.
Типичными примерами хромосомных болезней, связанных с нарушением количества хромосом, можно считать такие хромосомные синдромы, как синдром 47,ХY+Y, синдром 47,ХХ+Y и синдром Тернера-Шерешевского. Больные с этими синдромами жизнеспособны. Интеллект, как правило, не нарушен.
Полисомия Y, Синдром 47,ХY+Y встречается с частотой 1 : 1000 в нашей стране и 1 : 250 в США. Пол – мужской, рост высокий (средний рост – 186 см), имеют психопатические черты характера, эмоционально неустойчивы, агрессивны, склонны к асоциальному поведению, гомосексуализму. Добавочная Y-хромосома больше влияет на поведение, чем на интеллект, добавляет не дюжую физическую силу, иногда акромегалические черты (значительно развитая нижняя челюсть и лобные пазухи), неправильный прикус и расположение зубов, вальгусную давиацию суставов, патологию позвоночника (например, типа spina bifida), евнухоидное строение туловища, микроорхидизм или крипторхизм.
Примерно 80 % больных с этим синдромом имеют лёгкие признаки психического недоразвития с неравномерной своеобразной структурой интеллектуального дефекта. У этих больных в большей степени страдают предпосылки интеллектуальной деятельности, рано обнаруживаются дисгармония эмоционально-волевой сферы и формирование аномальных качеств личности при негрубом недоразвитии абстрактного мышления.
В раннем возрасте эти дети мало пользуются речью и обнаруживают признаки аутистического поведения. Они малообщительны, замкнуты, плохо сходятся с детьми, не проявляют глубоких привязанностей к близким людям. В школьном возрасте более отчётливо проявляются неустойчивость внимания, неусидчивость, неспособность к длительному интеллектуальному напряжению и целенаправленной трудовой деятельности. Эмоционально-волевые нарушения выражаются в беспричинных колебаниях настроения, взрывчатости, импульсивности и агрессивности по незначительному поводу. В тоже время больные внушаемы, легко имитируют поведение окружающих. Дети и подростки с синдромом 47,XY+Y при конфликтных ситуациях часто дают эксплозивные реакции с агрессией, совершают побеги из школы и дома. У некоторых больных отмечается наклонность к воровству, поджогам и другим правонарушениям. Эти дети и подростки могут легко усваивать программу вспомогательной школы, но их школьная и трудовая адаптация нарушена в связи с выраженной патологией поведения.
Мужчины фертильны, дети – нормальны, уровень андрогенов повышен, нормальные семьянины и работники. Мужчины с этим синдромом считаются самыми антисоциальными больными.
Среди маньяков, отличающихся серийностью своих преступлений, очень часто обнаруживаются больные с этим синдромом. Именно поэтому всех серийных убийц, насильников, садистов, душегубов, некрофилов, педофилов, герантофилов, зоофилов и прочих маньяков после задержания в первую очередь везут в психиатрическую клинику для проведения определения вменяемости и наличия лишней Y-хромосомы. В случае обнаружения добавочной хромосомы преступника из тюремной камеры переводят в палату № 6 (для буйных) психиатрической клиники и назначают пожизненное лечение, освобождая от наказания за их преступления.
Считается, что лишняя хромосома «заставляет» идти человека на преступление. Поэтому он включает весь свой интеллект на совершение преступления и сокрытие его следов. Доказано, что не все больные с этим синдромом совершают преступления. Практически всегда требуется внешний толчок, запускающий этот механизм. Больной не в состоянии бороться с желанием совершать преступления. Вид преступления зависит, как правило, от каких-то жизненных ситуаций, знаний, подсмотренных случаев, но всегда имеет тенденцию к серийности.
Около 20 % больных имеют нормальный или даже повышенный интеллект. Такие больные продумывают в мельчайших подробностях все детали преступления, они очень осторожны, бдительны, никогда не оставляют следов, улик, живых свидетелей и жертв живыми. Свой интеллект, высокий рост, физическую силу и угрожающие внешние данные больные эффективно используют с целью подавления психики жертвы, которые практически никогда не оказывают сопротивления, позволяя себя завести в любые недоступные другим людям места.
Больной с синдромом 47,ХY+Y был очень достоверно показан в знаменитом голливудском фильме «Молчание ягнят», где преступник-людоед имел высокое медицинское звание (доктор психологии Лептор) и помог следователю, не выходя из камеры, с помощью одного интеллекта отыскать серийного некрофила.
В действительности, больных с этим синдромом поймать за свои преступления очень трудно, так как они досконально изучают тактику и стратегию работы следователя, судмедэксперта, особенностей судопроизводства и т.д., то есть оказываются более подготовленными, чем обычный следователь. Поэтому просчитывают свои действия на шаг вперёд следователя.
Истинная частота синдрома 47,ХХ+Y не известна. Пол – женский, интеллект не ниже среднего, поведение в быту и на работе характеризуется всегда с лучшей стороны, могут иметь семью, дети – нормальные. Лишняя мужская хромосома придаёт этим женщинам дополнительные мужские четы: физическую силу, особенности телосложения (высокий рост, широкие плечи, более развитые мышцы).
Антисоциального поведения, в отличие от мужчин с добавочной Y-хромосомой, у женщин с этим синдромом не замечено. Как правило, из них получаются замечательные спортсменки. Однако, на спортивных соревнованиях высочайшего уровня (типа Олимпийские игры) всех спортсменок с мужскими чертами в обязательном порядке проверяют на применение мужских половых гормонов и их синтетических аналогов, а также на добавочную мужскую хромосому.
В случае обнаружения Y-хромосомы в кариотипе спортсменки её снимают с соревнований и не допускают соревноваться с женщинами, имеющими нормальный набор хромосом (46,ХХ). Так, что олимпийской чемпионкой среди женщин такая спортсменка, к сожалению, стать не может. Однако большинство из них могут гордо носить менее значимые спортивные звания и даже не знать об этой проблеме, так как, успешно занимаясь спортом, не достигли олимпийских вершин и не подвергались принудительному кариотипированию.
Синдром полисомии по Х-хромосоме (синдром 47,ХХ+Х, синдром 48,ХХ+ХХ и др.) встречается только у женщин. Значительных изменений фенотипа у них может и не быть вообще, так как лишние Х-хромосомы, как правило, спирализованы и представлены половым хроматином.
Такие больные могут иметь потомство, так как половина их гамет несёт нормальный набор хромосом. У больных может наблюдаться лёгкая умственная отсталость, причём, чем больше Х-хромосом в кариотипе, тем более выражены умственная отсталость, половой инфантилизм и неспецифические фенотипические изменения.
Обычно такие женщины высокого роста с искривлениями и деформациями позвоночника, пятнами депигментации на теле. На кончиках пальцев у них превалируют дуговые узоры, вследствие чего уменьшен дельтовый индекс и гребневой счёт. При варианте 49, ХХХХХ девочки мало жизнеспособны и обычно погибают в первые годы жизни.
С
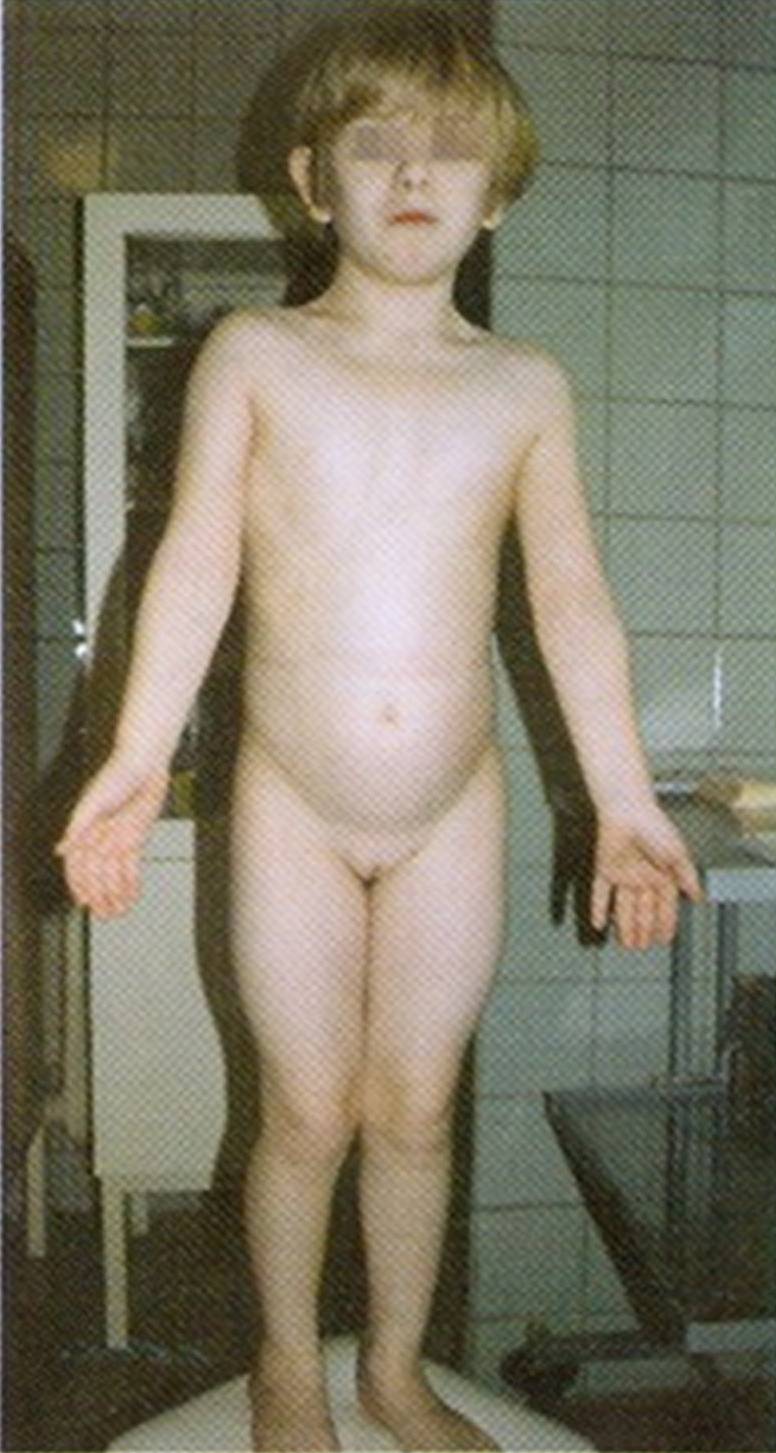
Это женщины низкого роста с половым инфантилизмом, недоразвитием наружных и внутренних половых органов (например, «слепое» узкое влагалище), недоразвитыми маткой и яичниками (больная 13 лет, рисунок 21). Яичники обычно не содержат развивающихся яйцеклеток, гипертрофирован клитор, отсутствуют вторичные половые признаки, молочные железы, соски недоразвиты и широко расставлены, ареолы втянуты, пигментация ареол слабая или вообще отсутствует, оволосение на лобке и под мышками отсутствует или очень слабое на лобке, месячных не бывает или они однократны, характерно бесплодие. Клиническими симптомами этого синдрома являются малый вес и рост уже при рождении, короткая шея, лимфэдема на кистях и стопах (припухлость), избыток кожи на шее (в виде складок), низкорасположенные деформированные ушные раковины, врождённые пороки сердца (коарктация аорты), эпикант, антимонголоидный разрез глаз, низко посаженные уши, низкий уровень волос, деформация грудной клетки, клинодактилия безымянного пальца, укорочен и искривлён мизинец, деформированы и глубоко посажены ногти, расширен угол atd, на кончиках пальцев преобладают круговые узоры, из-за чего увеличен гребневый счёт, широкие ладони, вальгусные колени, на коже много пигментных пятен, широкие плечи, узкий таз, короткие толстые ноги.
Нередко отмечается эпикант, а также микро- и ретрогнатия, узкое и высокое твёрдое нёбо, коарктация аорты, стеноз легочной артерии, не заращение межжелудочковой перегородки, подковообразная почка.
У этих женщин интеллект, как правило, не страдает, однако отмечается психический инфантилизм и эмоциональная лабильность. У больных резко снижено выделение эстрогенов и повышена экскреция гонадотропина.
На рентгенограммах трубчатых костей отмечается задержка окостенения, хотя рост таких женщин прекращается в 15 – 18 лет, а слияния эпифизов и метафизов нет даже в 25-летнем возрасте. Определяются увеличение медиальных мыщелков бедренных костей и уменьшение большеберцовых, истончение латеральных концов ключицы, остеопороз костей.
Диагноз синдрома Тернера-Шерешевского обычно устанавливается, когда наблюдается отставание девочки в росте и половой инфантилизм. Важным для диагностики этого синдрома является исследование полового хроматина в букальном эпителии. Если его там не обнаруживают, то это свидетельствует о моносомии по Х-хромосоме. Известно, что в клетках женского организма одна Х-хромосома в интерфазе неактивная, она спирализована и образует половой хроматин, или тельце Бара, которое обнаруживается у ядерной оболочки. У мужчин полового хроматина нет, так как у них одна Х-хромосома, которая функционально активна.
Начиная с 60-х годов ХХ века, генетики обратили внимание на наличие хромосомных мозаиков, у которых соматические клетки имеют неодинаковый набор хромосом в результате не расхождения хромосом на первом – втором делении после оплодотворения. В результате такого не расхождения возникают разнообразные мозаики, у которых часть клеток имеет, например, 47 хромосом, часть 45 хромосом.
Описаны мозаики типа XY/XO, XX/XO, XX/XXX, XXX/X и многие другие, а также мозаики, часть тела которых трисомна по хромосоме № 21, а часть нормальна. Однако аналогичные аномалии в распределении других хромосом, возникающие при первых делениях зиготы, практически всегда летальны.